





细胞焦亡是近年来发现并证实的一种新的程序性细胞死亡方式,其特征为依赖于炎性半胱天冬酶(主要是caspase-1,4,5,11),并伴有大量促炎症因子的释放。细胞焦亡的形态学特征、发生及调控机制等均不同于凋亡、坏死等其他细胞死亡方式。研究表明,细胞焦亡广泛参与感染性疾病、神经系统相关疾病和动脉粥样硬化性疾病等的发生发展,并发挥重要作用,对细胞焦亡的深入研究有助于认识其在相关疾病发生发展和转归中的作用,为临床防治提供新思路。
细胞焦亡属于程序性细胞死亡方式,包括焦亡在内的还有凋亡、自噬、坏死等,都是曾经或者目前的国自然热点,小优在之前写过很多相关的文章,直达链接如下:(文末相关阅读推荐)
虽然焦亡作为程序性死亡的热门话题已经持续了一段时间,但是大家对他的研究从未停止,尤其是其机制研究。NLRP3 炎症小体是引起细胞焦亡的一种经典途径,最近研究发现,通过WNK对细胞内离子浓度感知研究,可以调控NLRP3 炎症小体,进而调控细胞焦亡。
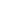
通过 WNK1 信号传导调节 NLRP3 激活和细胞焦亡的模型
在 NLRP3 炎症小体启动步骤(信号 1)之后,NLRP3 激活刺激(信号 2)诱导 K +和 Cl-从细胞流出,从而触发 NLRP3 炎症小体组装。WNK1 感应到细胞内 Cl-的减少并发生自磷酸化。WNK1磷酸化会介导其下游靶标 STK39/OXSR1发生磷酸化,后者激活阳离子Cl-协同转运蛋白以恢复离子浓度并抑制进一步的 Cl-流出。正常细胞内Cl-浓度的恢复进一步抑制NLRP3炎性--体激活和细胞焦亡。
NLRP3 炎症小体是一种细胞内多蛋白复合物,在暴露于病原体和组织损伤后,它会组装并产生炎症免疫反应。这种复合物主要存在于巨噬细胞中,由 NLRP3蛋白、ASC 衔接蛋白和 caspase-1 的前体 pro-caspase-1 组成。NLRP3 炎症小体是先天免疫中强大的参与者,可以抵御入侵的病原体和损伤,但 NLRP3 激活失调引起的过度炎症可归因于多种疾病,如炎症性肠病、动脉粥样硬化、类风湿性关节炎、痛风和阿尔茨海默病。因此,了解 NLRP3的调节机制对于制定对抗这种过度炎症的治疗策略至关重要。
不含赖氨酸 [K] (WNK) 丝氨酸/苏氨酸激酶是在所有哺乳动物中发现的激酶亚家族,其命名是因为它们在亚域 II 中缺乏催化赖氨酸,通常在激酶中保守以结合 ATP 。在渗透胁迫或细胞内低 Cl-期间,WNK 被激活并磷酸化效应激酶SPS/Ste20 相关富含脯氨酸丙氨酸激酶 (STK39) 和氧化应激反应 1 (OXSR1)。STK39 和 OXSR1 控制 SLC12 阳离子-Cl-协同转运蛋白家族的活性,从而调节细胞离子通量,即 Na+、K+和 Cl- 。WNK信号传导途径对于维持血压和肾脏稳态至关重要,WNK1 和 WNK4 的功能获得突变是导致多种高血压的原因。
最近的一项研究揭示了 WNK1 信号在通过胞吞作用去除凋亡细胞中的新作用。研究表明,WNK1 或其下游靶标 SLC12A2 协同转运蛋白的破坏导致凋亡细胞吸收显着增加,并在吞噬细胞中从抗炎反应转变为促炎反应,表明 WNK1 信号传导在抑制炎症中的作用。
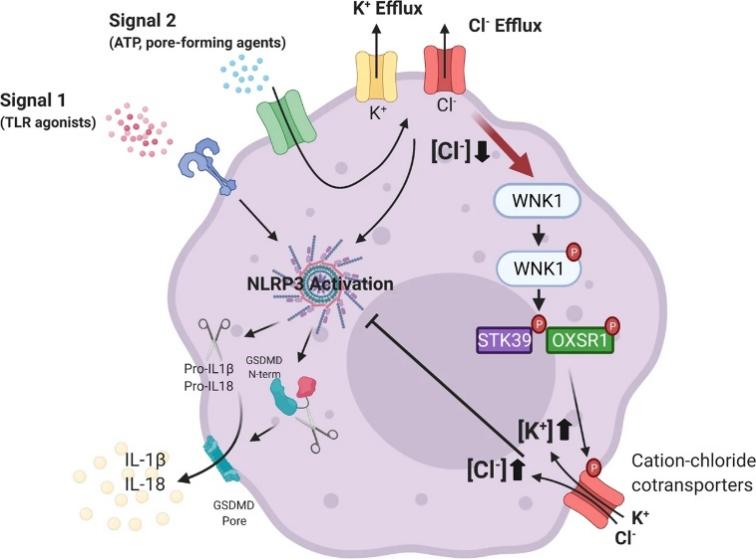
2005年-2021年 pyroptosis Pubmed 发表文献数统计
今天解读的文章是来自Nature的一篇2021年的高分文献,题为“Chloride sensing by WNK1 regulates NLRP3 inflammasome activation and pyroptosis”,该文章发现了细胞焦亡的一种新的研究思路。表明WNK1通过其对阳离子Cl- 浓度的调节,NLRP3炎性小体活化受到抑制,从而增加调控的另一层的NLRP3活化途径。话不多说,赶紧和小优一起解读文章吧,内容很多,全是干货,有需要的老师可以先收藏哦!
1. 抑制 WNK1 增加 NLRP3 炎症小体激活
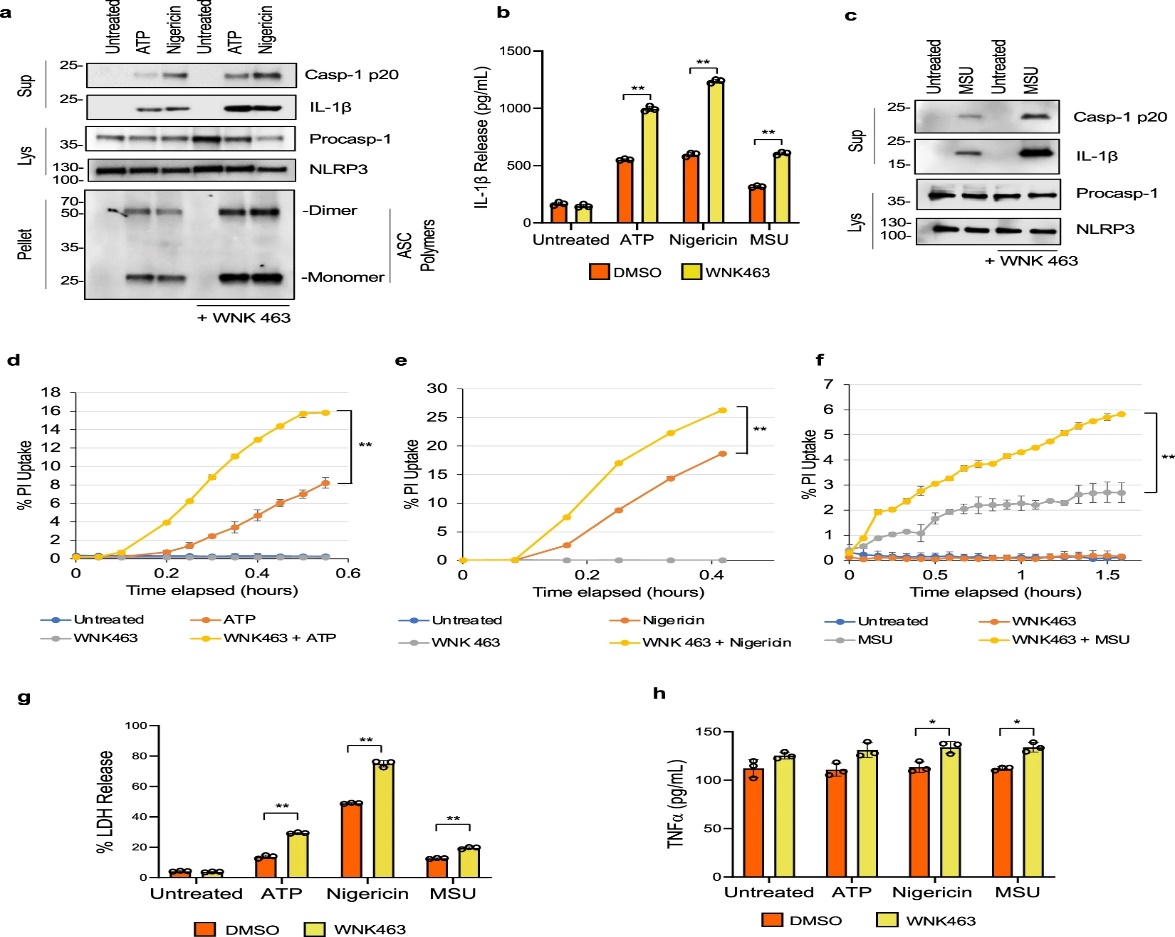
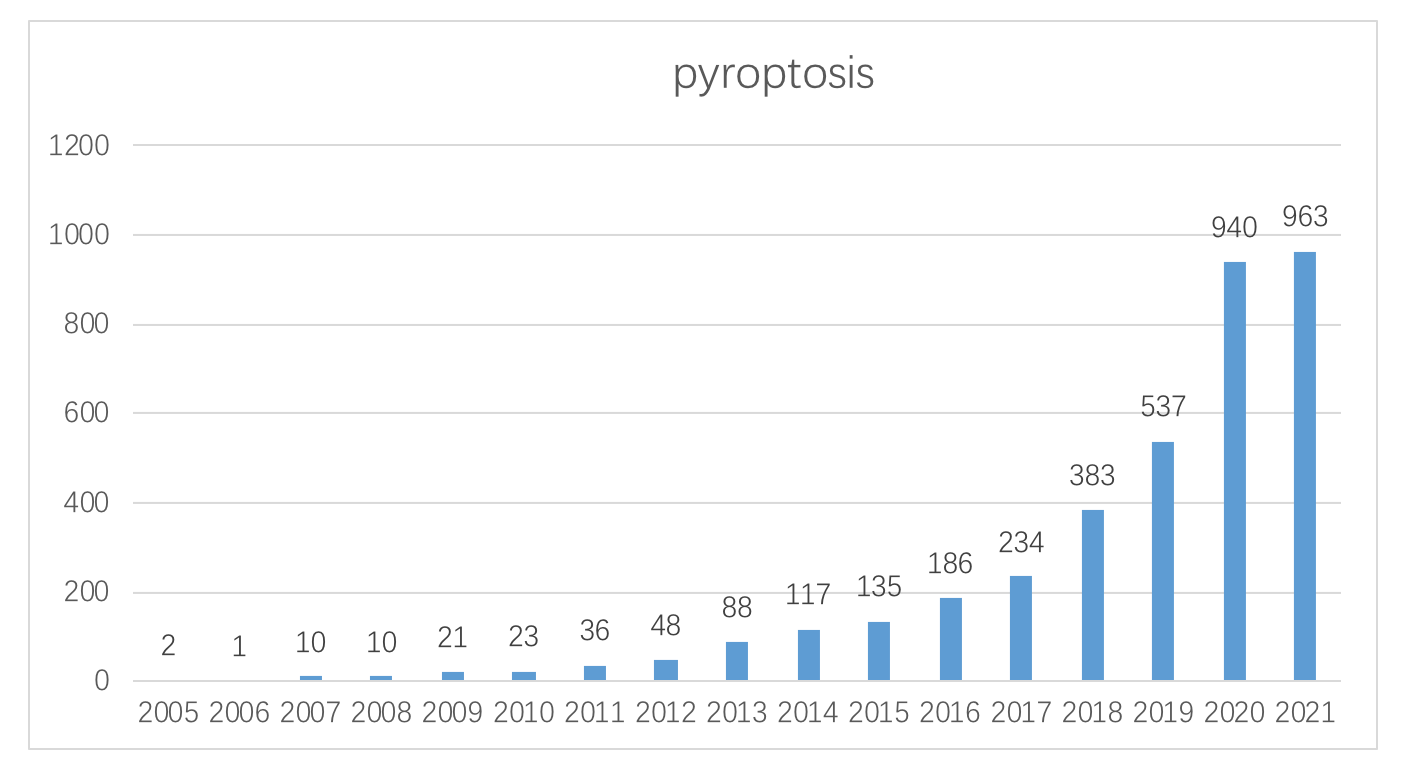
图1 WNK463 对 WNK 的抑制增加了巨噬细胞中 NLRP3 炎性体的激活。
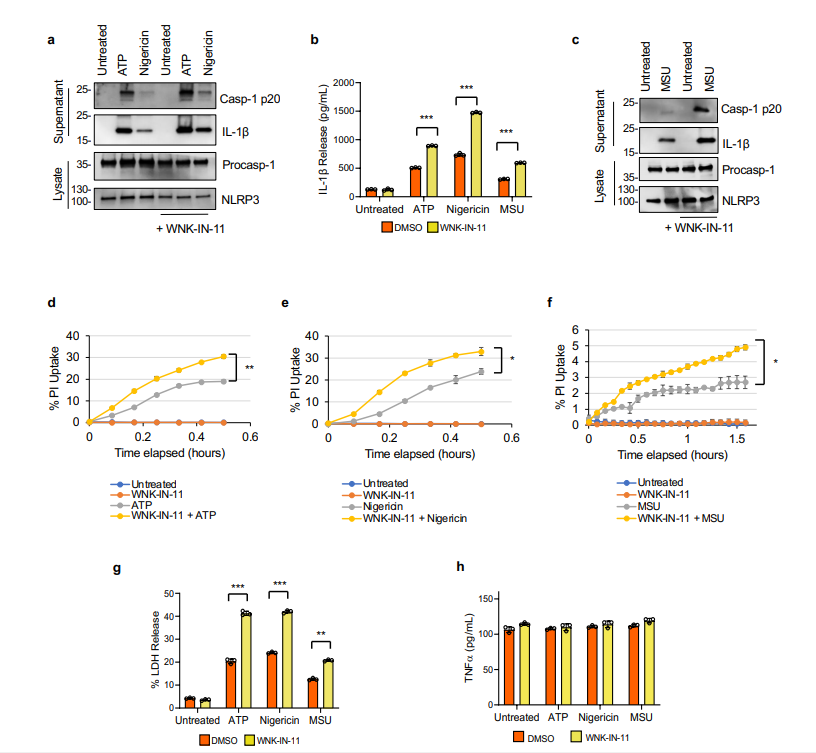
补充图1 WNK-IN-11对WNK1的抑制增加了巨噬细胞中NLRP3炎症体的激活
为了研究无赖氨酸 (WNK) 激酶是否是细胞内离子稳态和细胞体积的主要调节剂, 参与巨噬细胞中 NLRP3 激活的调节,作者评估了 WNK 激酶的药理学抑制对炎性体激活的影响。并用ATP,nigericin,或尿酸单钠(MSU)晶体处理LPS引发的原代骨髓源巨噬细胞(BMDM)在选择性使用WNK激酶的存在或不存在抑制剂WNK463 。文章中观察到 NLRP3 活化显着增加,这可以通过用 ATP(图 1a、b)、nigericin(图 1a、b)或 MSU(图 1b、c) 在 WNK463 存在下与对照处理相比。作者观察到在WNK463 存在下用 ATP、nigericin或 MSU 处理的细胞中碘化丙啶摄取增加(图 1d-f)以及乳酸脱氢酶(LDH)释放增加(图 1g)对照,表明焦亡增加。尽管 WNK463 单独处理,在没有炎症小体激活的情况下诱导了 TNF 产生的显着增加(图 1h),但炎症小体与 ATP、nigericin或 MSU 的激活并没有进一步增加 TNF 的产生。WNK463 对 TNF 的这种作用似乎对该药物具有特异性,并且当 BMDM 用 WNK1 抑制剂 11 处理或当它们的 WNK1 被删除时未观察到。
由于 WNK1 在大多数组织中表达,并且已显示通过调节 T 细胞活性在免疫系统中发挥作用,作者测试了最近开发的特异性 WNK1 抑制剂 11 (WNK-IN-11)对 NLRP3 炎症小体的影响在 LPS 引发的初级 BMDM 中激活。与 WNK463 类似,WNK-IN-11 增加了 NLRP3 炎性小体的激活,如用 ATP、nigericin或 MSU 处理的细胞培养上清液中的半胱天冬酶 1 激活和 IL-1β 产生所测量的(补充图 1a、b)。与对照细胞相比,WNK-IN-11 处理还增加了用 ATP、nigericin或 MSU 处理的 BMDM 中碘化丙啶的摄取和 LDH 释放(补充图 1d-g)。WNK-IN-11 对 TNF 的产生没有影响(补充图 1h)。结果表明 WNK1 在 NLRP3 炎症小体激活中起主要抑制作用,其抑制作用通过增加 NLRP3 寡聚化来增强炎症小体激活。
2. WNK1的条件性敲除增加了 NLRP3 炎症的某些激活
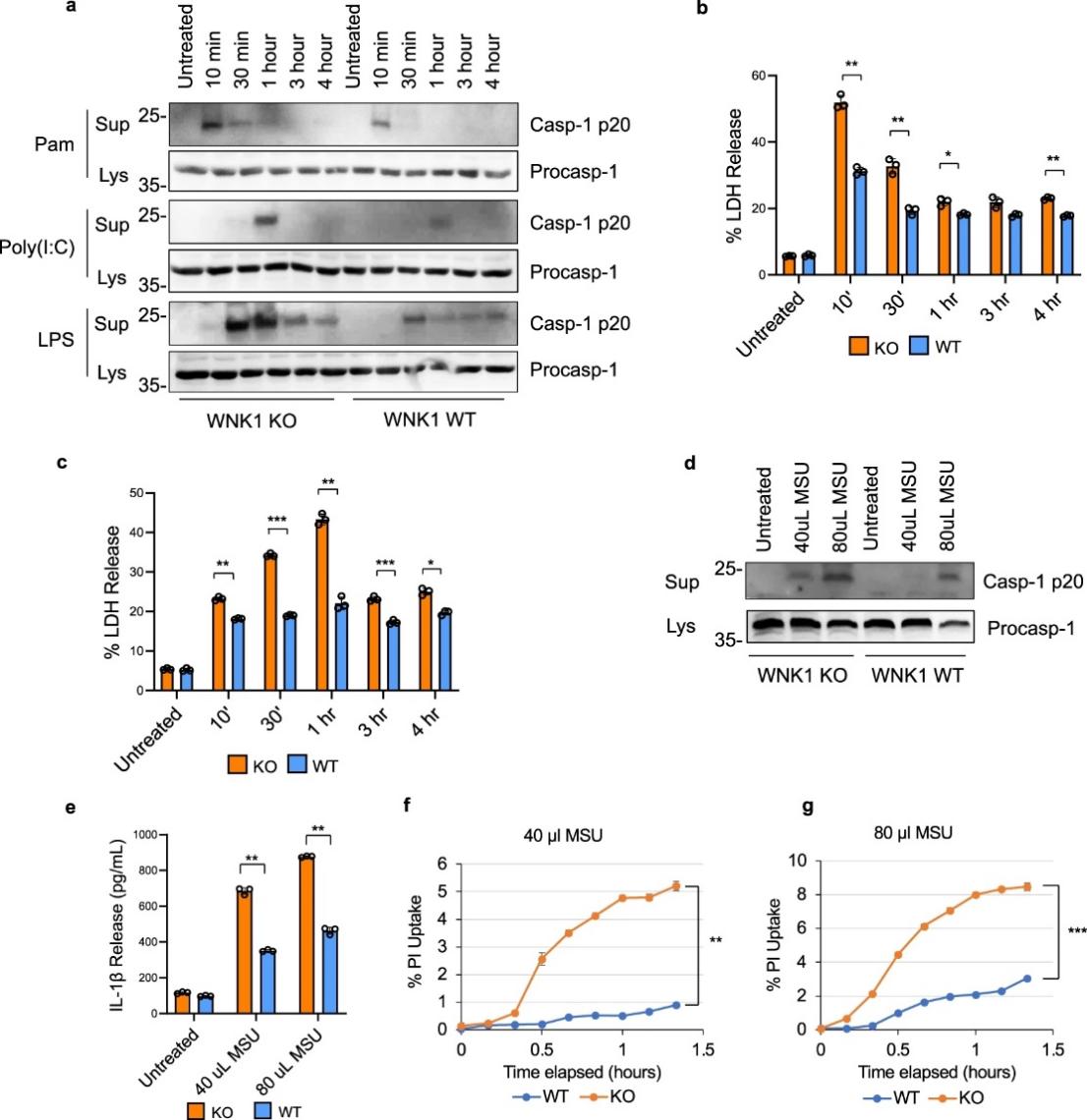
图 2:他莫昔芬诱导WNK1 敲除增加了巨噬细胞中 NLRP3 炎症小体的激活。
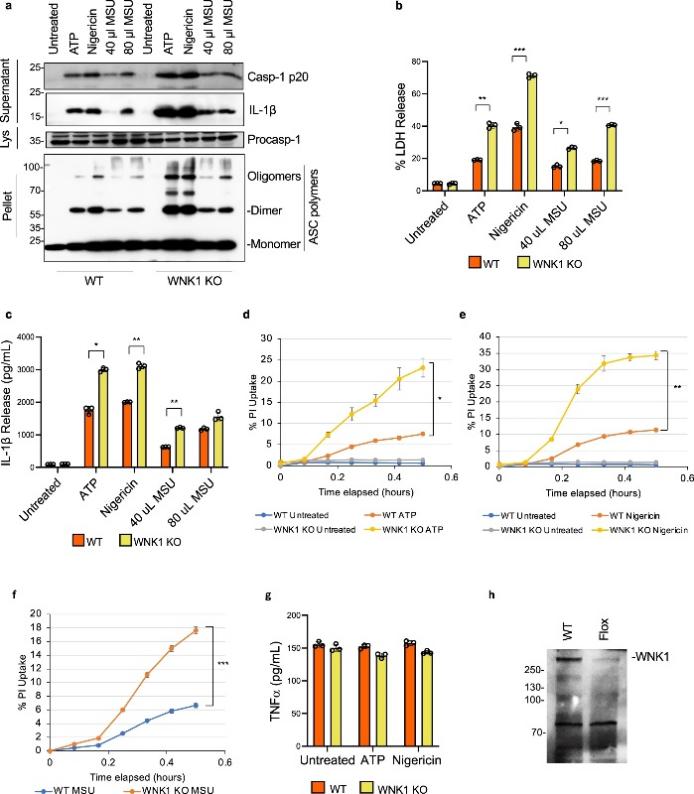
图 3:条件性 WNK1 敲除增加了巨噬细胞中 NLPR3 炎症小体的激活
为了评估条件性 WNK1 缺失对 NLRP3 炎症小体激活的影响,作者用 Pam3CSK4、Poly I:C 或 LPS引发了他莫昔芬处理的野生型和Wnk1 -/flox -CreERT2 +、 iBMDMs,然后是 ATP(图 2a)。观察到与对照他莫昔芬处理的野生型细胞相比,他莫昔芬诱导的 WNK1 敲除细胞中 NLRP3 活化显着增加。这表明 WNK1 缺失不影响 NLRP3 启动步骤,但它增强了 NLRP3 激活步骤。这种增加与 LDH 释放增加有关(图 2b、c),表明细胞焦亡增加。类似地,caspase 1 激活增加(图 2d)、IL-1β 释放(图 2e)和 PI 摄取(图 2f、g)) 在用 MSU刺激他莫昔芬处理和 LPS 引发的Wnk1 -/flox -CreERT2 + iBMDMs 后,以剂量依赖性方式出现。在原代 Wnk1 -/flox -CreERT2 + BMDM(未显示)中观察到类似的结果。总之,这些结果验证了WNK 抑制剂数据,并强调了 WNK1 在调节 NLRP3 炎症小体激活中的重要性。
为了在不使用他莫昔芬的情况下开发一个模型并允许在体内评估 WNK1 缺乏对 NLRP3 炎症小体激活的影响,作者生成了携带在骨髓特异性 Cre 重组酶 LysMCre 控制下的floxed Wnk1基因的小鼠。这些小鼠仅在完全分化的巨噬细胞中删除Wnk1,而不会干扰心血管发育。WNK1条件性敲除小鼠 ( Wnk1 flox/flox LysMCre + ) 存活下来并且在表型上与它们的野生型同胞没有区别。从这些小鼠中分离的 BMDM 用 LPS 引发并用 ATP、nigericin或 MSU 处理并分析 caspase 1 激活和 ASC 寡聚化(图 3a)、LDH 释放(图 3b))、IL-1β 释放(图 3a、c)和 PI 摄取(图 3d-f)。与WNK1 抑制剂和他莫昔芬诱导的Wnk1缺失的结果一致,作者观察到条件 WNK1 KO BMDM 中 NLRP3 激活和细胞焦亡与其野生型对照相比显着增加(图 3a-f)。总的来说,这些数据进一步验证了 WNK1 在抑制巨噬细胞中 NLRP3 炎症小体激活中的关键作用。
3. WNK1/OXSR1/STK39 信号通路调节 NLRP3 的激活
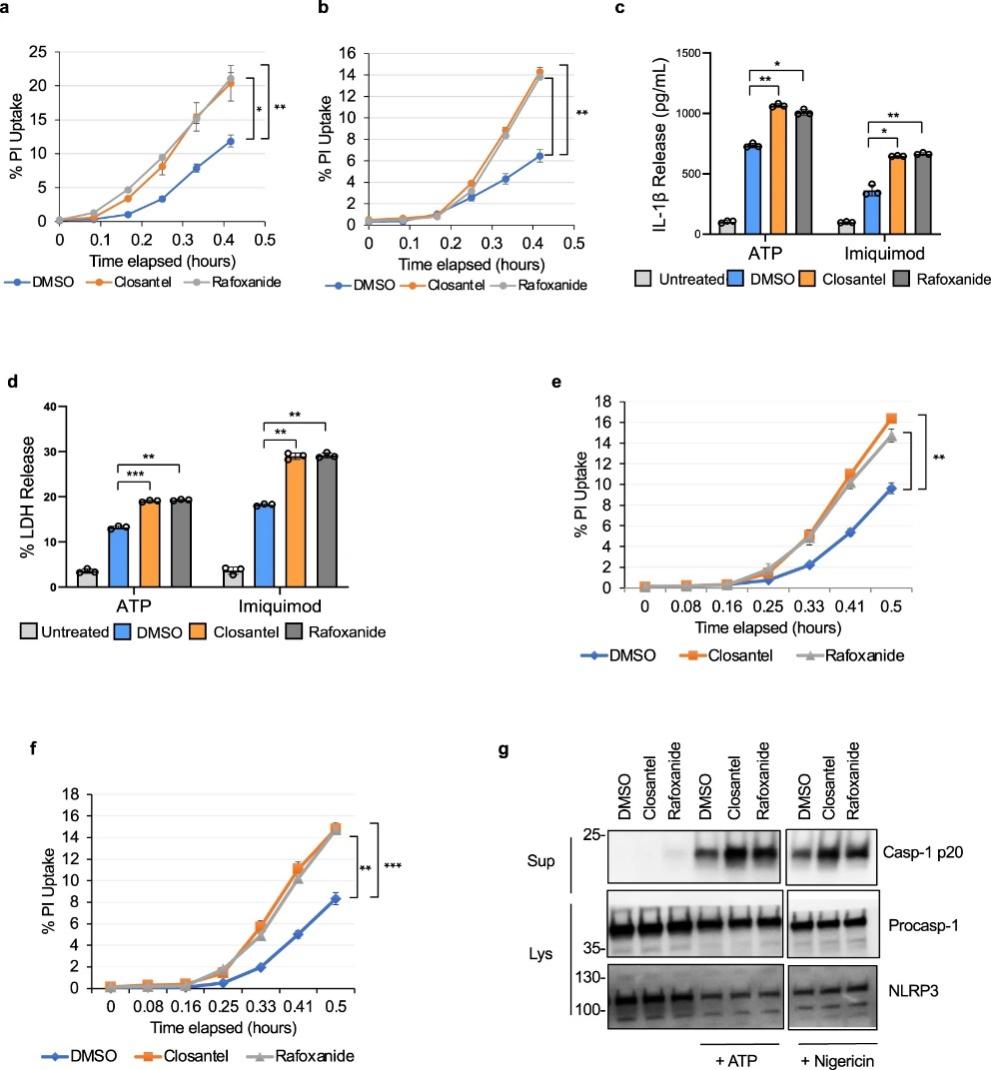
图 4:STK39/OXSR1 的药理学抑制增加了巨噬细胞中 NLRP3 炎症小体的激活
WNK1 通过激活氧化应激反应 1 (OXSR1) 和 STE20/SPS1 相关的富含脯氨酸/丙氨酸 (STK39) 激酶来调节细胞内离子浓度,这些激酶通过作用于质膜Na+来调节细胞内Na+、K+和Cl-离子通量/ K+、Cl-共转运蛋白。考虑到Cl-和 K +流出在 NLRP3 激活中的重要作用,我们研究了 WNK1 对 NLRP3 激活的调节是否依赖于其典型的 OXSR1/STK39 信号通路。我们使用了两种不同的抑制剂,Rafoxanide 和 Closantel,它们通过结合到 STK39 和 OXSR1 激酶的 C 末端结构域上的变构位点来抑制它们。在用 ATP(图4a)或咪喹莫特(图 4b)刺激 NLRP3 之前,用 Closantel 或 Rafoxanide 预处理 LPS 引发的原代野生型 BMDM 10 m 导致与载体对照相比更多的碘化丙啶摄取。这些细胞还显示出更多的 IL-1β 产生(图 4c)和 LDH 释放(图 4d)) 控制。当在永生化的野生型 BMDM 中测试这些抑制剂时,获得了类似的结果(图 4e-g)。总之,这些结果表明规范 WNK1/OXSR1/STK39 激酶信号传导是控制 NLRP3 炎症小体激活所必需的。
4. WNK1 通过Cl -传感机制调节 NLRP3 的激活
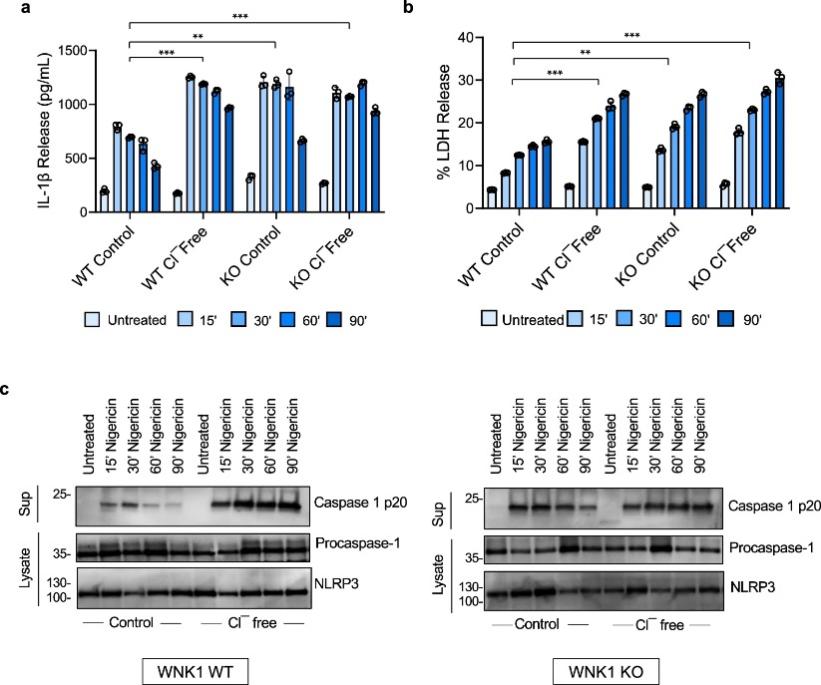
图 5:WNK1 通过 Cl -感应机制调节 NLRP3 炎症小体激活
WNK1 已被证明可以直接感知细胞内低 Cl -浓度,导致其激活。一旦被激活,WNK1 会磷酸化OXSR1 和 STK39 激酶,进而磷酸化并激活SLC12A2 (NKCC1)、SLC12A1 (NKCC2) 和 SLC12A3 (NCC) Na+,K+和Cl-共转运蛋白以维持细胞内正常的Na+,K+和Cl-离子浓度。WNK1 通过在检测到细胞内 Cl-下降时激活阳离子Cl-协同转运蛋白来抑制 NLRP3 激活的假设。激活的协同转运蛋白通过介导Cl-和K+从细胞外介质流入来抵消Cl-和 K+损失。因此,WNK1将无法抵消氯-和K +时,野生型的BMDM刺激在Cl-损失-不含介质,因为阳离子Cl-共转运有氯的绝对要求-为他们的活动。LPS引发的野生型的BMDM表明显著更高IL-1β的生产,LDH释放和胱天蛋白酶-1激活(图 5A-C)当与nigericin在含Cl刺激相对于对照介质free介质。此外,LPS引发的缺陷WNK1的BMDM,其预期具有没有协同转运蛋白活性,在nigericin的刺激free介质引起IL-1β的生产没有增加,LDH释放或胱天蛋白酶-1激活高于在可见正常对照培养基(图 5)。事实上,炎性活化通过在WNK1 KO细胞的IL-1β和LDH释放所量化的水平类似于在那些在氯的野生型细胞中看到- -free媒体(图 如图 5a、b ) 所示,表明与正常培养基中的野生型 BMDM 相比,WNK1-KO 中 NLRP3 炎性体的激活增加是由于 WNK1 缺失导致共转运蛋白活性受到抑制。这些结果表明,通过在无Cl-培养基中孵育来抑制协同转运蛋白活性可以对NLRP3活化产生与WNK1的缺失/抑制相同的作用。
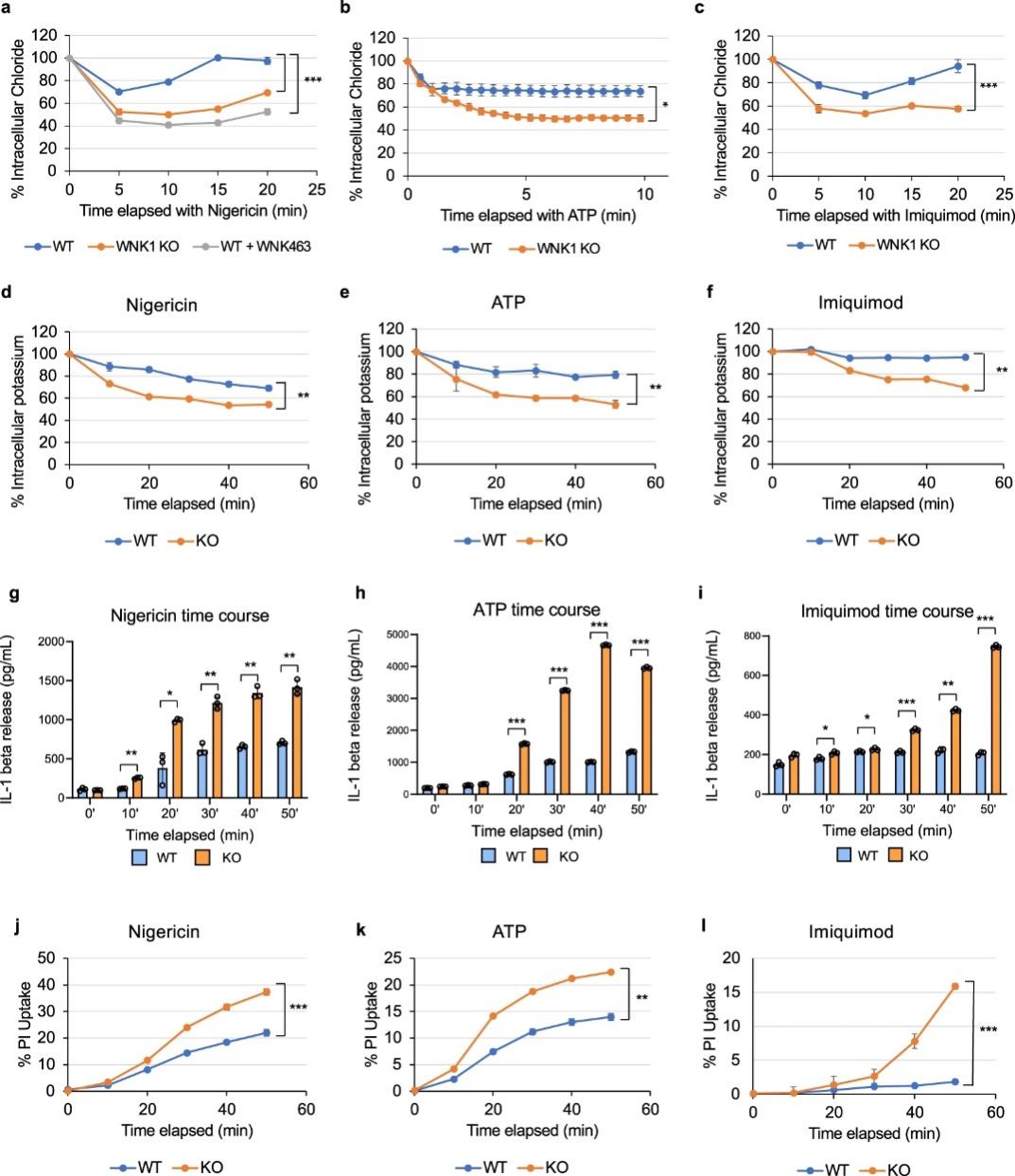
图 6:WNK1 调节巨噬细胞中的细胞内 Cl –
阳离子Cl-协同转运蛋白在 WNK1 介导的 NLRP3 激活调节中的重要性通过使用 MQAE(一种荧光 Cl-离子指示剂)测量细胞内 Cl-得到进一步验证。在静息状态下,野生型和条件性 WNK1 敲除 BMDM 具有相似的细胞内 Cl-水平。当用 NLRP3 刺激物nigericin、ATP 或咪喹莫特而非 AIM2 炎症小体激活剂 dA:dT 处理时,与 WNK1 KO BMDM 相比,野生型 BMDM 的细胞内Cl-损失显着降低(图 6a-c,)。野生型 BMDMs在nigericin或 ATP 处理后 5 分钟损失约 25% 的细胞内Cl-(图 6a、b),在咪喹莫特处理后 10 分钟损失约 30% 的细胞内Cl-(图 6c))。相比之下,条件性 WNK1 KO 细胞以及用 WNK463 处理的野生型 BMDM在 5 到 10 分钟内失去了约 50% 的Cl-(图 6a-c)。NLRP3 刺激对细胞内Cl-水平的影响与 NLRP3 状态无关,因为在存在或不存在 WNK1 抑制剂的情况下用nigericin处理的 NLRP3 敲除 BMDM 显示出类似的Cl-模式在类似处理的野生型 BMDM 中观察到的损失。由于WNK1通过阳离子Cl-协同转运蛋白调节的K+通量与Cl-通量偶联,与用相同NLRP3刺激的WT对照相比,WNK1-KO BMDM中的细胞内K+损失也更大刺激(图 6d-f)。这些结果表明,如果没有 WNK1,BMDM 无法在 NLRP3 激活期间抵消其细胞内 Cl-和 K+ 的损失,因为它们无法调节其阳离子Cl-协同转运蛋白,这解释了增加的 NLRP3 激活和细胞焦亡(图 6g-l)。
5. K+和Cl-外排驱动 WT 和 WNK1 KO 细胞中的 NLRP3 激活
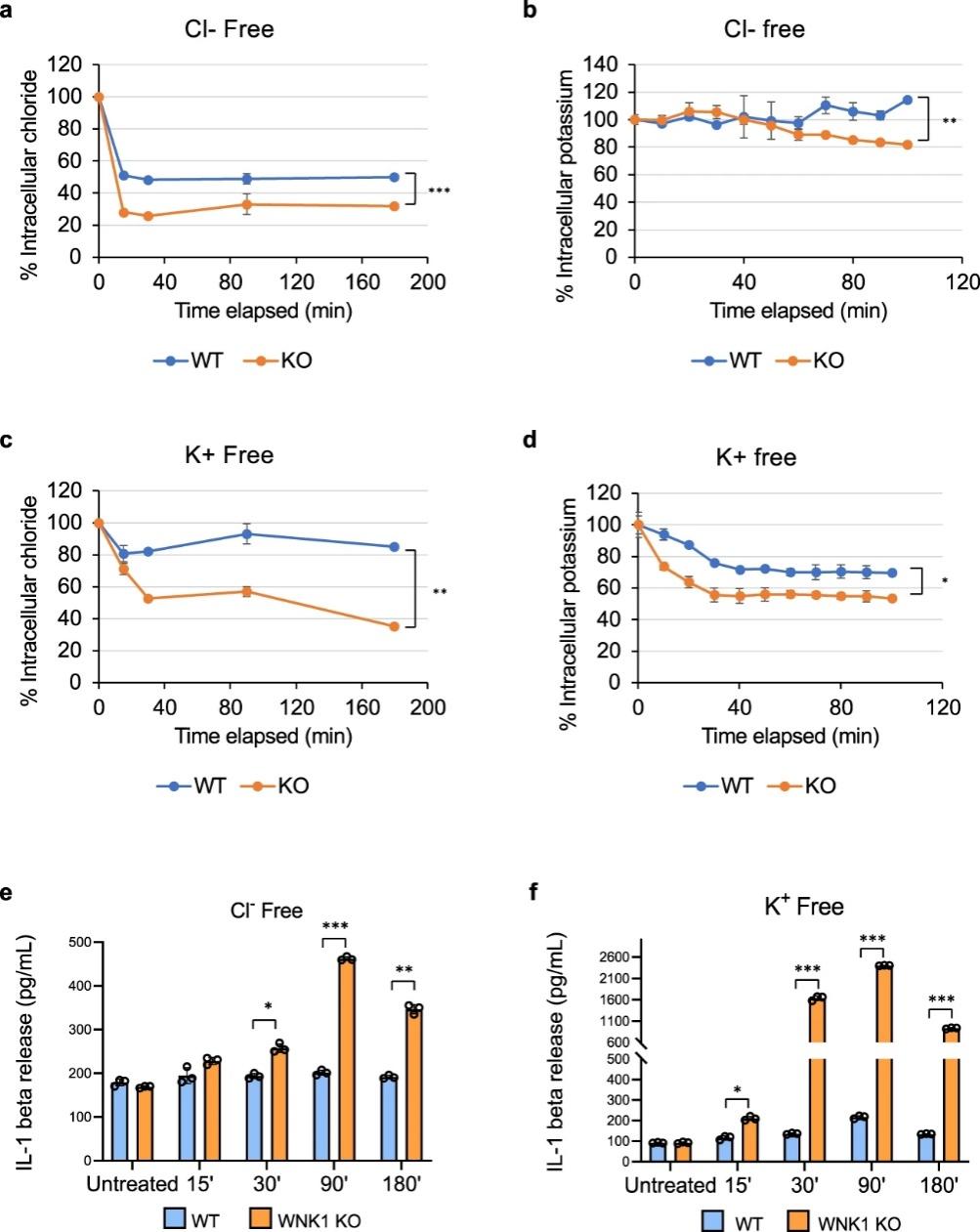
图 7:钾和Cl-流出驱动 WT 和 WNK-KO 细胞中的 NLRP3 激活
为了研究K+或Cl-流出是否促进 WNK1 缺陷型 BMDM 中 NLRP3 的激活,作者研究了了在无K+或无Cl-培养基中培养 BMDM 是否可以在没有外源性 NLRP3 刺激的情况下激活 NLRP3 炎性体。 在无Cl-培养基中孵育导致WT和WNK1-KO细胞中细胞内Cl-的快速下降(图 7a)。然而,在 WT 和 WNK1-KO 细胞中,在孵育的前 50 分钟内,细胞内K+的水平变化很小或没有变化(图 7b)。然而,细胞内K+的水平在较晚的时间点仅在 WNK1-KO 细胞中适度下降(图 7b)。在无K+培养基中孵育导致WT细胞中细胞内Cl-的小幅下降和K +的适度下降,但在WNK1-KO细胞中观察到细胞内Cl-和K +的更大下降(图7c, d )。对于在无Cl-培养基中的WT 细胞,在细胞内 K +没有类似下降的情况下,细胞内Cl-的下降(图 7a,b)没有激活 NLRP3 炎性体,如通过 IL-1β 分泌所测量的(图 7e))。相比之下,在无Cl-培养基中WNK1 -KO细胞中细胞内Cl-的下降和细胞内K+水平的最终小幅下降(图 7a、b)导致NLRP3活化(图 7e)。无K+培养基中K+和 Cl-水平的下降激活了 WT 和 WNK1-KO 细胞中的 NLRP3 炎性体,但 WNK1-KO 细胞中的 NLRP3 激活要大得多(图 7c、d、f)。这些结果表明Cl-和K+流出都驱动NLRP3 激活,并且这些通量受WNK1 调节。
6. WNK1在体内调节先天免疫反应
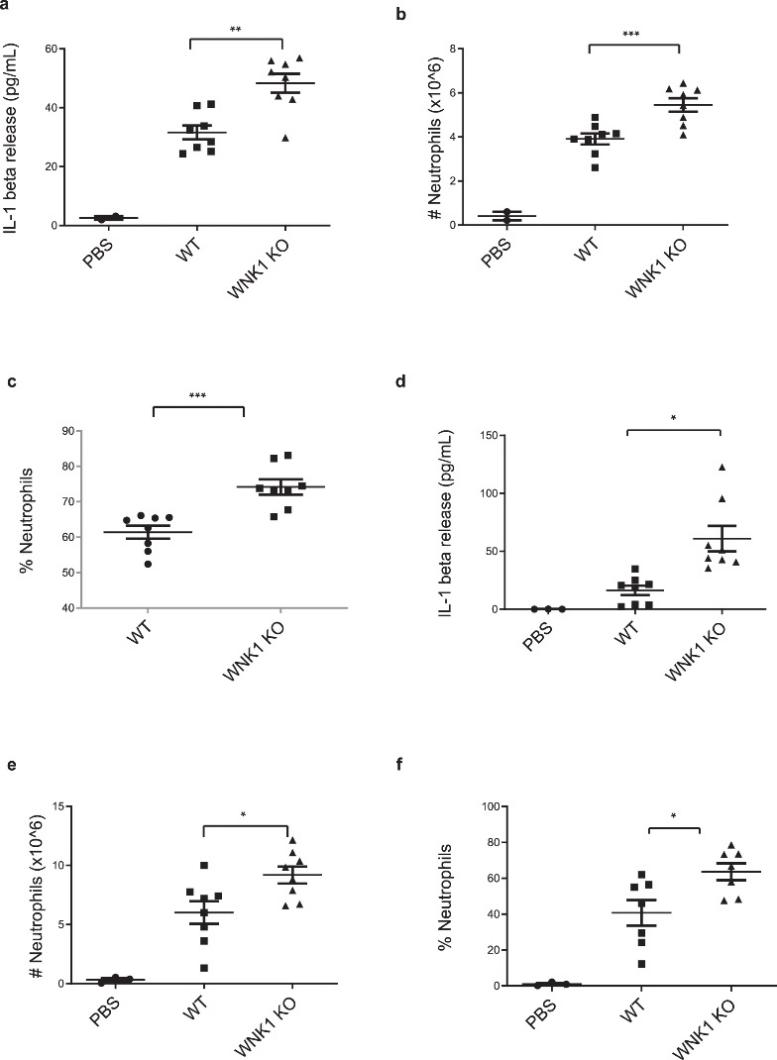
图 8:巨噬细胞中的 WNK1 敲除增加了体内 NLRP3 炎症小体的激活
为了证明体外发现的生理学相关性,作者研究了 WNK1 在 NLRP3 依赖性先天免疫反应中的作用。腹腔注射单钠尿酸盐 (MSU) 晶体会启动 NLRP3 依赖性免疫反应,其特征是中性粒细胞浸润和 IL-1β 释放到腹膜,研究员注射了 8-12 周大的Wnk1 flox/flox LysMCre +小鼠和Wnk1 +/+ LysMCre +在收集和分析腹膜渗出液之前,用 1 mg MSU 对 NLRP3 进行 6 小时“短”激活或 16 小时“长”激活。发现,在 MSU 处理 6 小时后,条件性 WNK1 KO 小鼠的腹腔渗出液中的 IL-1β 水平显着高于野生型同窝小鼠(图 8a)。渗出液的 FACs 分析揭示了条件 WNK1 KO 小鼠中浸润性中性粒细胞数量的增加(图 8b)以及中性粒细胞群体百分比的增加(图 8c)。在 MSU 处理 16 小时后,结果观察到 IL-1β 释放(图8d)、浸润的中性粒细胞数量(图8e)和渗出液群体中的中性粒细胞百分比(图 8d)增加 。 (图 8f ) 在条件 WNK1 KO 小鼠中与其野生型同窝小鼠相比。在短期或长期 MSU 治疗后,我们没有观察到 TNFα 水平的显着差异。总之,这些结果表明 WNK1 在体内 NLRP3 激活中起重要作用。
7. WNK4 不会调节 NLRP3 激活
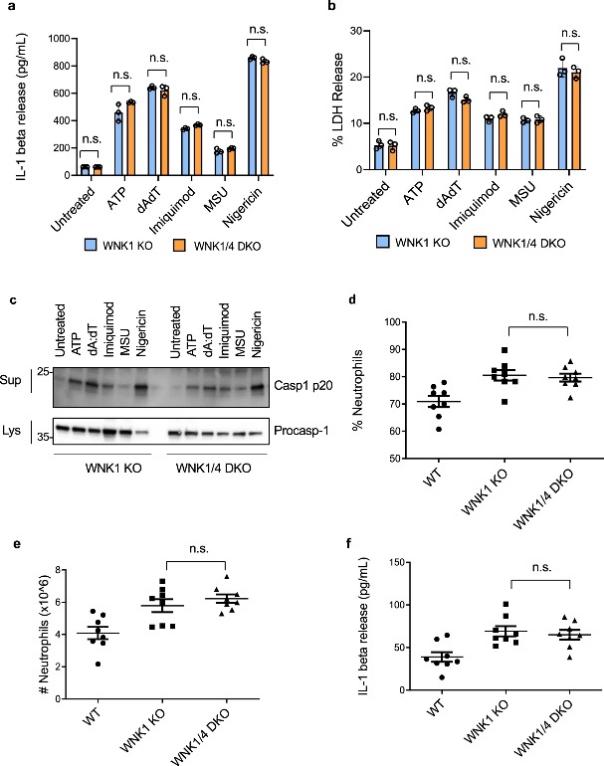
图 9:WNK4 敲除不会增加体外或体内 NLRP3 炎症小体的激活
WNK1和 WNK4都是Cl-敏感性激酶,因此作者研究了WNK4 是否像 WNK1一样增强炎症小体激活,或者 WNK4 是否对 WNK1 起补偿作用,WNK1/WNK4 双敲除会产生更多激活。与野生型相比,作者没有观察到 WNK4 KO BMDM 中 NLRP3 激活的增加。由于Wnk4敲除在小鼠中不是致命的,当通过 IL-1β 释放(图 9a)、LDH 释放(图 9b)或半胱天冬酶 1 激活(图 9c ) 当受到多种炎性体刺激时。在测量 MSU 注射后的免疫反应时,作者也没有看到体内 WNK1 KO 和 WNK1/4 DKO 之间的可观察差异,如渗出液中的中性粒细胞百分比(图 9d)、浸润到腹膜的中性粒细胞数量(图 9e),或 IL-1β 释放(图 9f)。这些结果表明 WNK1 而不是 WNK4 在调节 NLRP3 激活和免疫反应中至关重要。此外,WNK4 活性不能替代 WNK1 的丢失,因为与有条件的 WNK1 KO 相比,WNK1/4 DKO 没有显示出显着影响。
总结:
上述文章强调了 Cl -调节在 NLRP3 炎症小体激活中的重要性,通过检测巨噬细胞中的细胞内低Cl-水平,WNK1通过激活阳离子Cl-协同转运蛋白来平衡细胞内离子(Na+、K+和 Cl-),从而抑制 NLRP3 炎症小体的激活,从而防止过度炎症。
本文重点在于首次证明尽管小分子咪喹莫特被认为通过 K+ 独立机制,NLRP3 炎症小体的激活对 WNK1 缺失或抑制敏感,表明离子(K +和 Cl-)稳态在咪喹莫特诱导的 NLRP3 激活中起关键作用。
最后,研究表明了抑制 WNK1 通路的临床意义,包括使用利尿剂治疗高血压,因为可能会出现与 WNK1 通路调节 NLRP3 相关的炎症并发症。
本文涉及部分相关产品:
|
货号 |
产品名称 |
|
WNK1 Antibody |
|
|
IL-1β (3A6) Mouse mAb |
|
|
Caspase-1 (E2Z1C) Rabbit mAb |
|
|
Mouse Reactive Inflammasome Antibody Sampler Kit |
|
|
NLRP3 (D4D8T) Rabbit mAb |
|
|
ASC/TMS1 (D2W8U) Rabbit mAb (Mouse Specific) |
|
|
Anti-mouse IgG, HRP-linked Antibody |
|
|
Mouse IL-1 beta/IL-1F2 Quantikine ELISA Kit |
|
|
Mouse TNF-alpha Quantikine ELISA Kit |
|
|
WNK463 |
|
|
Nigericin |
|
|
WNK-IN-11 |
|
|
Rafoxanide |
|
|
Closantel |
|
|
N-Acetyl-L-cysteine |
|
|
Closantel |
|
|
广谱型蛋白酶抑制剂混合物 |
|
|
RIPA裂解液 |
|
|
Adenosine 5'-triphosphate disodium salt hydrate Grade I, >=99%, from microbial |
|
|
Propidium iodide >=94.0% (HPLC) |
|
|
STK39 |
|
|
SML2247 |
|
|
Imiquimod (50 mg) |
|
|
CytoTox 96® Non-Radioactive Cytotoxicity Assay |

