





当组织细胞中的氧气不充足,或供小于需时,即为缺氧。
早在上世纪90年代,Semenza就发现缺氧诱导因子(HIF1)是缺氧的重要调控因子。同时,Kaelin发现了Von Hippel-Lindau 蛋白 (VHL)参与缺氧反应的调控,随后,Ratcliffe发现Von Hippel-Lindau 蛋白 (VHL)通过泛素化参与HIF1α的降解。为表彰这三位科学家在低氧感应方向做出的杰出贡献,2019年授予他们诺贝尔生理学或医学奖。
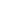
近20年来关于缺氧研究的文章呈线性增长,尤其是2019年来更是增长迅速。
2001-2022年PubMed 缺氧相关文献数
想必大家也更想对缺氧领域进行深入了解,今天小优也埋头苦读了几天几夜整理了几篇高分文献分别对缺氧的调控机制、与其他相关领域研究关联以及研究思路三个方面进行了详细阐述,篇幅有点长,干货满满,建议收藏后再研读哦!
缺氧调控机制
缺氧会激活相关信号通路,该通路主要受缺氧诱导因子 (HIF) 稳定性控制。在常氧条件下,HIF-α 亚基的脯氨酸残基被氧依赖性脯氨酰-4-羟化酶 (PHD) 羟基化。 Von Hippel-Lindau 蛋白 (VHL) 是一种 E3 泛素连接酶,与羟基化的 HIF-α 结合并作为 E3 泛素连接酶复合物的底物识别组分,导致 HIF 蛋白降解。HIF-α 亚基的天冬酰胺残基也被抑制 HIF 的因子羟基化(FIHs),它抑制 HIF 与共激活剂 p300/CREB结合蛋白的结合。缺氧状态下,PHDs 和 FIHs 的活性受到抑制,HIF-α亚基易位进入细胞核与 HIF 1β(HIF1B)结合形成异源二聚体,异二聚体 HIF-α:HIF-1β 转录因子复合物然后定位到靶基因的缺氧反应元件 (HRE),导致相关基因转录上调。这些基因涉及细胞存活、增殖、运动、代谢、pH 调节、细胞外基质功能、炎性细胞募集和血管生成。

HIF作为一个相对上游的事件,通过影响下游通路靶点,进而影响不同疾病的发生。
缺氧与其他相关领域研究的关系
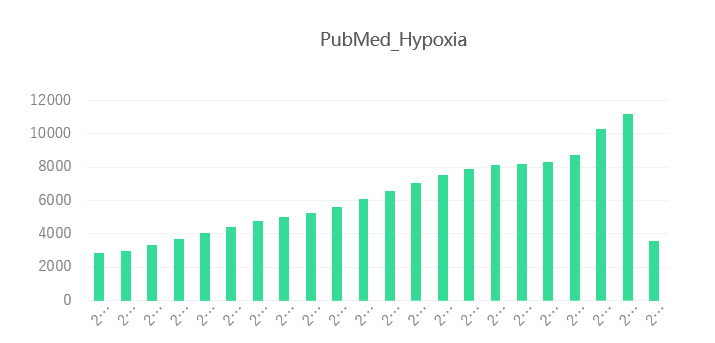
缺氧与EMTHIF-1α 可诱导多个 EMT诱导转录因子。在前列腺癌和肝细胞癌中证明,缺氧条件下HIF-1α 驱动 Wnt/β-catenin对 EMT 的诱导; HIF1α 还可直接与 TWIST 的启动子(一种 EMT 的诱导剂)中存在的缺氧反应元件 (HRE) 结合,并在乳腺癌细胞中激活EMT。在 COX-2 和 NF-KB 存在下,HIF-1α 介导的癌症相关成纤维细胞 (CAF) 的激活已被证明以 ROS 依赖性方式诱导前列腺癌细胞中的 EMT。此外,在肾癌和肝癌细胞中发现,HIF-1α 通过抑制 E-钙粘蛋白或上调 SNAIL 和 ZEB1诱导不同程度的 EMT 。
缺氧与炎症
在包括慢性炎症和缺血组织多种病理免疫环境中,缺氧和炎症是存在的伴随事件,
在这些病理状态下,组织缺氧导致缺氧诱导因子 (HIF) 途径的激活,核因子-κB (NF-κB) 通路除了在免疫的转录调节和控制免疫细胞功能的方面起作用。此外, NF-κB 被炎症刺激物(如细胞因子或细菌产物)激活,也表现出对缺氧的敏感性;缺氧通过这种机制调节病理免疫生态位中的炎症基因表达。此外,HIF 和 NF-κB 通路之间存在广泛的串扰。例如,由细胞因子或细菌产物驱动的经典 NF-κB 活性增加会促进 HIF1α mRNA 的转录,从而在慢性炎症条件下促进 HIF 活性;其他因组织炎症而升高的信号传导介质,也调节免疫细胞中的 HIF 活性,从而有助于调节免疫和炎症。

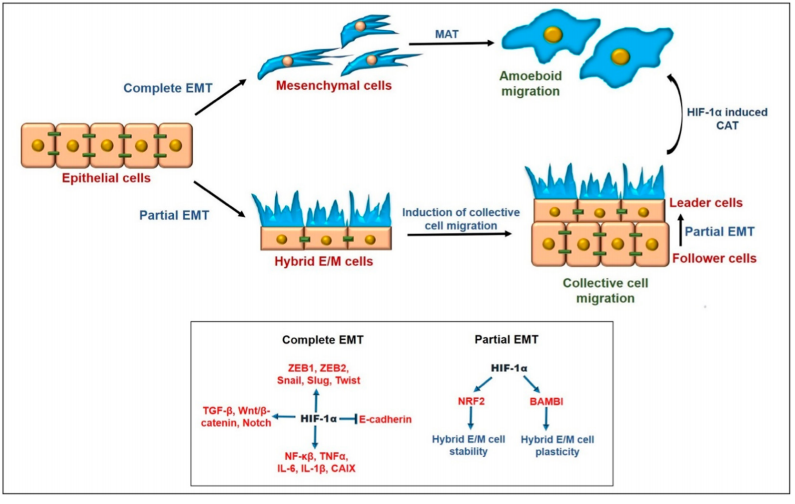
缺氧与代谢
缺氧和HIF信号通路以多种方式影响内皮细胞的功能和血管生成。一方面,缺氧诱导血管生成基因的转录激活,同时也在转录后调节促血管生成趋化因子和受体,从而促进内皮细胞迁移到血管生成部位。而且,VEGF等血管生成生长因子在缺氧时进一步稳定可增加内皮细胞中的糖酵解通量。此外,缺氧刺激内皮细胞增殖和发芽,并通过细胞外基质重塑促进血管重塑。在人肝癌和视网膜色素上皮细胞系中观察到,血管内皮细胞(ECs )在缺氧时可能通过 HIF 稳定作用上调 PFKFB3酶,进一步增强对内皮细胞增殖和血管生成的影响。缺氧除了对ECs的直接影响外,癌细胞的缺氧还可以向ECs发出 信号,从而改变其代谢。乳酸在(缺氧)肿瘤环境中的积累可导 致ECs摄取乳酸,随后转化为丙酮酸进行进一步氧化。
缺氧与神经退行性疾病
遗传性阿尔茨海默病(AD)危险因素与β淀粉样蛋白斑块相关小胶质细胞减少相关,缺氧中HIF1α和HIF1靶基因的上调,减少β淀粉样蛋白斑块相关小胶质细胞的覆盖范围,进而损害了小胶质细胞的活动。当这种情况再加上其他系统性疾病(如:心血管疾病)导致的大脑供氧减少时,小胶质细胞就无法提供保护,与疾病相关的病理就会增加。
缺氧与基因组稳定性
RNA聚合酶II (Pol II)在非缺氧条件下启动许多HIF靶基因的转录,但在大约30-60个核苷酸后暂停,需要HIF-1结合释放。在低氧乳腺癌细胞中,HIF-1α 通过组装多蛋白复合物响应缺氧触发释放暂停的 Pol II,HIF-1招募TRIM28和具有催化活性的dna依赖蛋白激酶(DNA-PK)到HREs,释放暂停的Pol II,从而刺激转录产物的延伸。

文献解读
缺氧条件下葡萄糖剥夺在肝细胞癌上皮间质转化中的关键作用

1 人肝癌标本中缺氧的存在以及GLUT1、Ki67和EMT相关靶点的表达情况

图1 基于18F-FDG摄取的HCC样本中HIF1α、GLUT1、CD31和EMT相关蛋白表达的差异。
高摄取FDG的HCC患者的HCC组织中肿瘤区域的hif1-α表达和膜状GLUT1阳性区域明显增多。在高摄取FDG的HCC患者中,CD31表达无变化,但肿瘤区域血管分布不均匀。Ki67的表达与emt相关蛋白N-cadherin或vimentin的表达呈负相关。结果表明,EMT相关蛋白,如N-cadherin或vimentin,在HCC癌细胞中根据缺氧条件下的增殖速率而有差异表达。
2 缺氧敏感HepG2细胞在缺氧条件下增殖和EMT的失调。
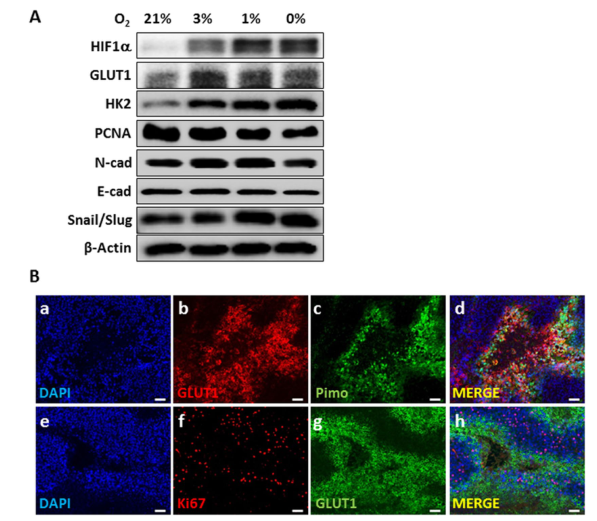
图2 缺氧相关蛋白在HepG2细胞中的表达
3 缺氧对hepg2异种移植模型中Ki67、GLUT1和EMT表达的影响

图3GLUT1在坏死区的表达
在hepg2异种移植模型中,观察到在缺氧区GLUT1表达上调,并且在一些坏死区连续表达(图3A)。本研究将HepG2细胞在零氧无氧环境下培养3天。缺氧1 d后,hif1 α表达增加,而随着缺氧培养时间的延长,hf1α的表达减少,GLUT1的表达增加。因此,在坏死区GLUT1的表达是阳性的( 图 3B , C )。通过体外结合实验测定了放射标记的葡萄糖在HepG2细胞中的摄取,在缺氧状态下,放射性葡萄糖的摄取持续了3天(图3D )。因此,GLUT1的表达可能不是诱导EMT过程的一个充分标志。
4 缺氧缺糖条件下EMT相关蛋白的协同表达。
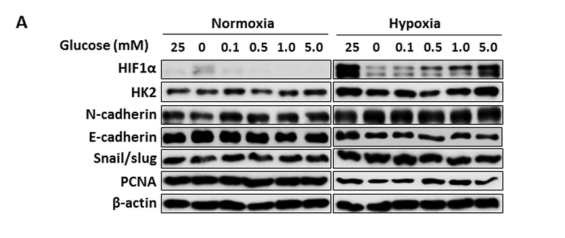
图4 缺氧缺糖后EMT相关蛋白表达增加
在体外研究了缺氧条件下葡萄糖和缺氧对EMT相关蛋白表达的协同作用。如预期的那样,在1%氧浓度下,严重的糖剥夺可增加N-cadherin和Snail/Slug的表达,表明糖剥夺和缺氧协同增加EMT相关蛋白。
5 缺氧和增殖条件下EMT相关蛋白的协同表达。

图5 低氧条件下低增殖细胞EMT相关蛋白表达增加
有趣的是,与高糖HCC细胞相比,葡萄糖浓度为0 mM和0.1 mM的HCC细胞中PCNA含量下降(图4A);在HepG2和Hep3B细胞中敲除PCNA后,在完全缺糖的缺氧条件下,N-cadherin和Snail/Slug的表达显著增加(图5A)。因此,HCC细胞的增殖在诱导EMT过程中也起着重要作用。Ki67的表达随距坏死区的距离而变化(图5B)。与体外实验相似,观Ki67与N-cadherin或vimentin的表达呈负相关。因此,缺氧性条件下的葡萄糖剥夺可有效地调节体内EMT相关蛋白。本研究揭示了一个独特的微环境,包括缺糖、缺氧和低增殖率诱导EMT过程。
意义:本研究发现,EMT过程非常复杂,是由缺氧、细胞增殖低和葡萄糖扩散不良共同引起的。 这些发现在 PET 分析期间的临床实践中可能非常有用,可用于预测转移的预后和治疗结果。
缺氧通过HIF1破坏阿尔茨海默病小胶质细胞线粒体代谢

1 HIF1介导的应激反应途径在A β AM中被诱导-
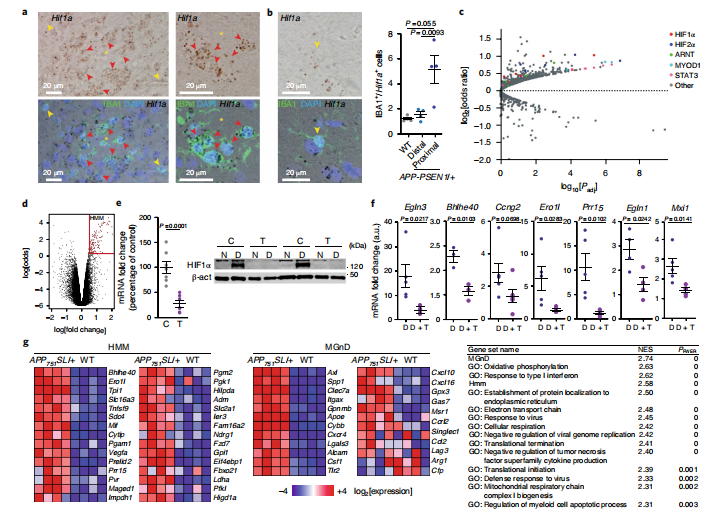
图1 HIF1介导的转录在AβAM中被激活
AD小鼠模型中发现Hif1a mRNA在Aβ斑块周围表达(图1a )。采用转录因子富集分析发现,HIF1 α和HIF2 α是小胶质细胞转录调控因子( 图 1c )的前两个蛋白,表明HIF介导的转录在β淀粉样蛋白斑块相关小胶质细胞(Aβ AM)中发挥着重要作用。通过差异表达基因进行主成分分析,确定差异表达基因(图1d),并在原代小胶质细胞培养中使用诱导型Cx3cr1 Cre::ERT2介导的Hif1a缺失证明HIF1对其进行调节(图1e,f)。
2 AD小胶质细胞氧化磷酸化(OXPHOS)相关转录增加。
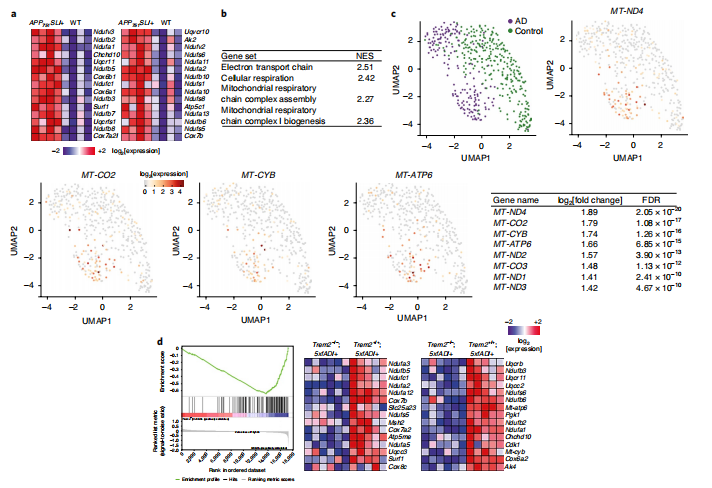
图2 AD小胶质细胞增加有氧呼吸相关转录
在APP和TAU神经退行性小鼠模型中,OXPHOS基因组显著富集,包括上调所有线粒体电子传递链复合物(复合物Ⅰ-Ⅳ)和复合物V( ATP酶) 编码蛋白的基因m RNA水平。这些数据被其他与有氧呼吸和ATP生成相关的基因富集所证实(图2b)。为了证实本研究结果也与人类疾病相关,参考最近一项人类单细胞RNA测序研究的数据。从死后人类AD样本中分离的小胶质细胞与对照小胶质细胞分析,编码OXPHOS的基因在AD样本的小胶质细胞中显著过表达(图2c)。
而且发现TREM2缺乏确实与5xfAD / +小鼠模型小胶质细胞OXPHOS基因的显著下调有关(图2d ),在AD小鼠模型和人类AD小胶质细胞中,有氧呼吸相关基因的转录以trem2依赖的方式增加。
3 Aβ AM线粒体具有延伸的特征
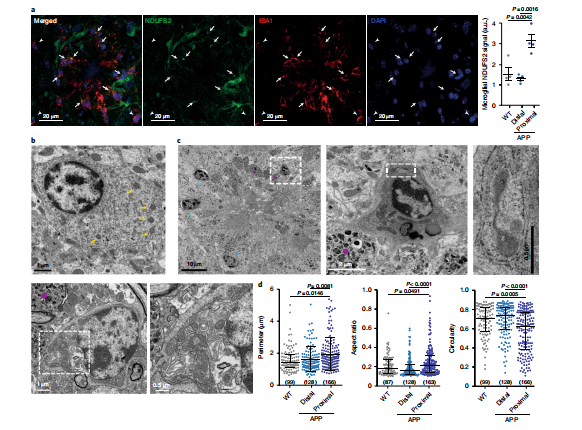
图3 在AD小鼠模型中线粒体被延伸
通过免疫荧光评估线粒体水平,观察到与野生型小胶质细胞或Aβ斑块远端小胶质细胞相比,NDUFS2复合物Ⅰ蛋白在AβAM中明显上调(图3a )。电子显微镜观察了AβAM线粒体的形态。远离Aβ斑块的小胶质细胞呈圆形线粒体(图3b ),而AβAM呈粗面内质网复盖的长形线粒体(图3c,3d);。这些结果表明,有氧呼吸是神经退行性DAM的共同特征,同时HIF1介导的基因表达的激活和线粒体的伸长提示Aβ AM代谢受损。
4. HIF1的过度激活引起小胶质细胞的静止
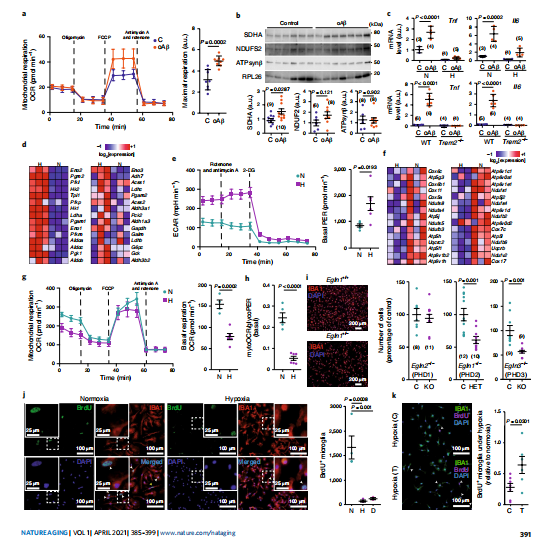
图4缺氧通过细胞内小胶质细胞HIF1诱导细胞周期阻滞
无论是体内、体外,线粒体活性通过oAβ处理上调。为了表征缺氧在小胶质细胞中的作用,首先分析了从暴露于低氧水平,缺氧诱导了一种强劲的转录反应,其特征是糖酵解基因的协同诱导(图4d)。与此相对应的是,在缺氧条件下,原代小胶质细胞的糖酵解率明显增加(图4e),低氧水平抑制线粒体OXPHOS(图4f-g)。
缺氧24和48小时导致细胞周期急剧停滞。为了区分缺氧诱导衰老(不可逆细胞周期阻滞)或静止(可逆细胞周期阻滞),将BV2缺氧培养物暴露于复氧24小时。有趣的是,细胞周期在常氧培养后完全恢复。为了证实HIF在缺氧介导的细胞周期阻滞中的作用,在缺乏PHD3或半剂量PHD2的情况下,小胶质细胞数量都有所减少(图4i),并且使用缺氧条件下有条件删除Hif1a的原代小胶质细胞培养(图1e)。如预期的那样,缺氧使BrdU+细胞数量减少约60%。然而,在缺乏HIF1α的培养物中,小胶质细胞增殖几乎完全恢复(图4k),这表明HIF1有助于缺氧条件下可逆的小胶质细胞周期阻滞。
5.通过HIF1降低线粒体代谢可降低AβAM
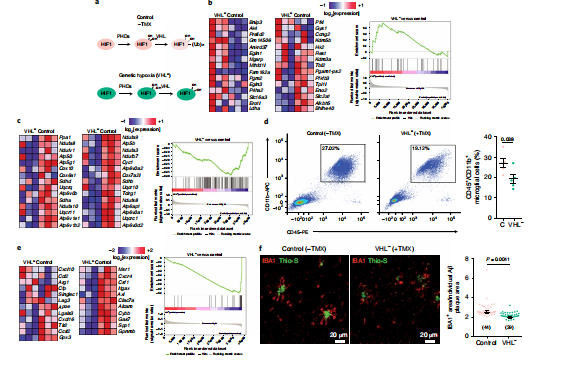
图5 HIF1的过度稳定降低了体内的AβAM
VHL缺乏在体内诱导了HMM,(图5b),OXPHOS基因组转录下降(图5c)。流式细胞术观察到,这种转录调控与小胶质细胞百分比的下降有关(图5d)。值得注意的是,VHL缺失诱导了共同小胶质标志(MGnD基因集;图5e),提示a β am减少。因此,通过定量a β斑块周围的IBA1免疫反应性,并观察到在无VHL时,皮质a β沉积的小胶质覆盖减少(图5f)。总之,这些结果表明,通过稳定HIF1下调a β am有氧呼吸可诱导小胶质细胞功能失调表型。
6.持续缺氧可增强Aβ局部病理。
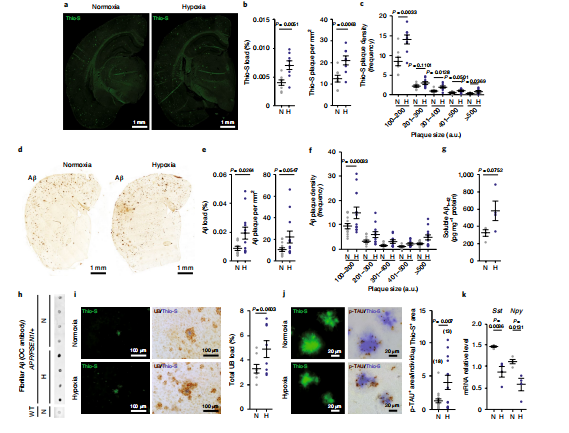
图6 系统性持续缺氧增强Aβ聚集、扩散和Aβ斑块相关的轴突营养不良
APP-PSEN1/+小鼠在缺氧和常氧状态下皮层Thio-S+和总Aβ+斑块的大小分布显示在缺氧状态下斑块富集,提示低氧促进Aβ聚集,导致更多新形成的斑块(图6a-f)。ELISA法检测常氧和低氧APP - PSEN1 / +小鼠血清中可溶性Aβ1 - 42的含量,发现在低氧条件下,可溶性Aβ1 - 42的含量有增加的趋势(图6g )总之,这些数据表明,持续的缺氧增强了Aβ在脑实质的聚集和沉积。
APP-PSEN1/+小鼠暴露于持续缺氧后显示泛素负荷增加的趋势(图7i),每个硫代硫+斑块p-TAU+营养不良神经突密度明显增加(图6j),生长抑素(Sst)和神经肽Y (Npy)的mRNA水平在持续缺氧应激下进一步显著降低(图6k)。因此,持续缺氧导致可溶性Aβ纤维寡聚物和新形成的致密核Aβ斑块的增加,并加重Aβ斑块相关的神经退行性现象。
7.缺氧脑区高病理的裸Aβ斑块
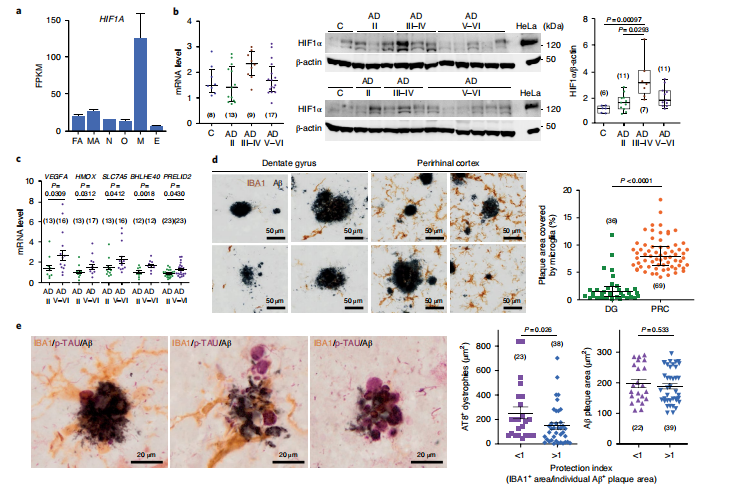
图7 人类易缺氧的大脑区域含有裸体Aβ斑块,局部轴突营养不良增加
与其他细胞类型相比,人小胶质细胞中HIF1a的转录本非常丰富(图7a ),mRNA (无明显趋势)和蛋白水平在AD病理中均上调。还发现AD ( Braak V-Ⅵ)人海马样品中多个HIF调节基因的mRNA水平上调(图7c ),提示病理晚期与诱导HIF1有关。与来自同一个体的瘤周皮质的斑块相比,齿状回分子层Braak V-Ⅵ期出现了明显的老年斑小胶质细胞脱群现象(图7d ),提示局部缺氧也启动了人AD脑的AβAM功能障碍,生成了裸Aβ斑块。
意义:本研究结果也为寻找能够改善小胶质细胞线粒体代谢适应Aβ斑块的药物铺平了道路,并可能减少AD的进展。
部分相关产品
| 货号 | 产品名称 |
| 13110S | PCNA (D3H8P) XP® Rabbit mAb |
| 13116S | N-Cadherin (D4R1H) XP® Rabbit mAb |
| 5741S | Vimentin (D21H3) XP® Rabbit mAb |
| 13116S | N-Cadherin (D4R1H) XP® Rabbit mAb |
| 3528S | CD31 (PECAM-1) (89C2) Mouse mAb |
| abs42016321-25mg | DAPI |
| abs9331 | Trizol |
| 612519 | ATP Synthase Bta Pure 10 150ug |
| G3893 | Monoclonal Anti-Glial Fibrillary Acidic Protein (GFAP) antibody |
| 5292S | BrdU (Bu20a) Mouse mAb |
| 36169S | HIF-1α (D1S7W) XP® Rabbit mAb |
| NB100-105 | Mouse Monoclonal HIF-1 alpha Antibody (H1alpha67) |
| 9449S | Ki-67 (8D5) Mouse mAb |
| 3700S | β-Actin (8H10D10) Mouse mAb |
| 17198S | Iba1/AIF-1 (E4O4W) XP® Rabbit mAb |
| NA931 | MOUSE IGG HRP LINKED WHOLE AB |
| MA526534 | NDUFS2 Monoclonal Antibody |
| 11998S | SDHA (D6J9M) XP® Rabbit mAb |
| DAB142 | hA beta (aa1-42) Qki (1 Kit) |
| DAB140B | hA beta (aa1-40) Qki (1 Kit) |
| SH30809.01 | RPMI 1640 |
| SH30042.01 | Trypsin |
| SV30010 | Penicillin |
| SH30406.05 | FBS |
| Abs972 | 胎牛血清(优级) |

