




Silencing Apoe with divalent-siRNAs improves amyloid burden and activates immune response pathways in Alzheimer's disease
APOE4与阿尔茨海默病:功能获得性机制确立新靶点
作为阿尔茨海默病(AD)最强的遗传风险因子,载脂蛋白E4(APOE ε4)的致病机理长期存在争议:其风险效应究竟源于蛋白本身的毒性功能(功能获得性机制),还是因氨基酸位点变异导致APOE4功能缺陷(功能缺失性机制)?这一科学问题直接决定了AD治疗策略的本质差异——若为前者,干预需靶向抑制APOE4表达;若为后者,则需通过增强APOE4功能实现保护。尽管啮齿类模型研究呈现两种假说的并存局面,但人体数据始终是验证病理机制的金标准。
近期,斯坦福大学Michael D. Greicius教授团队在《Neuron》期刊发表突破性研究,通过人类基因组学分析为这场争论画上阶段性句点。研究团队从近6万例全基因组测序数据中,精准识别出7例携带APOE功能缺失型突变的健康个体,并对其展开深度表型分析。结果显示:这些APOE蛋白活性完全缺失的受试者,其AD核心病理标志物——脑脊液Aβ42/Aβ40比值及磷酸化tau蛋白(p-tau)水平均处于正常范围,且临床随访中未见认知衰退或神经影像学异常。该发现以人体证据强有力支持APOE4通过功能获得性机制驱动AD病理进程,即其致病性源于异常功能活性而非单纯功能不足。
这一结论为AD治疗策略带来革命性启示:靶向APOE4的沉默或基因编辑技术(如CRISPR-Cas9介导的等位基因特异性敲除)或成为延缓AD进展的新方向。研究同时揭示,APOE基因型与AD病理的关联存在剂量效应依赖性——携带1个APOE4等位基因即可使AD发病风险提升3-4倍,而纯合突变体风险更呈指数级增长。这种精确的机制阐释,不仅填补了AD遗传学领域的关键认知空白,更为开发针对APOE信号通路的精准疗法奠定了科学基础。

APOE功能研究突破:人体数据破解阿尔茨海默病遗传机制之谜
在遗传学研究领域,基于人体的功能验证始终是揭示基因致病机制的金标准。然而,载脂蛋白E(APOE)功能缺失型变异因其人群极端罕见性(等位基因频率<0.01%),长期制约着相关致病机理的解析。尽管十年前曾有病例报告记载1例APOE完全缺失个体的临床特征,但单一病例无法建立变异与阿尔茨海默病(AD)风险的因果关联。
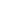
为突破这一研究瓶颈,斯坦福大学研究团队系统性开展了人类遗传学筛查。通过整合阿尔茨海默病测序计划(ADSP)全外显子组测序数据库(n=36,361)与全基因组测序数据库(n=20,503),研究人员采用深度变异解读流程,成功鉴定出7例携带APOE功能缺失型变异的受试者(图示)。其中包括6例单核苷酸变异(SNV)携带者(受试者1-6)及1例结构性变异(SV)导致APOE完全缺失的个体(受试者7)。
该队列呈现三大特征:
- 变异类型多样性:涵盖移码突变(p.Leu28Profs3)、无义突变(p.Gln160)及外显子4缺失(c.344-2A>C)等多种致病变异形式;
- 临床表型异质性:7例受试者年龄跨度42-78岁,其中6例认知功能正常,1例仅表现主观认知下降(未达MCI诊断标准);
- 病理标志物阴性:所有个体脑脊液Aβ42/Aβ40比值、总tau蛋白(t-tau)及磷酸化tau181(p-tau181)水平均处于正常范围,且氟代脱氧葡萄糖-PET(FDG-PET)显示代谢模式无AD特征性改变。
这种罕见遗传学队列的构建,为解析APOE致病机制提供了关键人体证据。研究显示,尽管APOE蛋白功能完全丧失,但受试者未出现AD病理表型,从而否定了"功能缺失性保护"假说,转而支持APOE4通过毒性功能获得驱动AD病理进程的机制模型。这一发现不仅填补了AD遗传学领域的重要认知空白,更为开发针对APOE信号通路的精准干预策略(如ASO介导的APOE4特异性沉默)奠定了科学基础。
受试者1:APOE4失能个体的病理悖论与保护性暗示
1号受试者为男性,终年90岁,携带APOE3/4杂合基因型,其APOE4等位基因因移码突变(p.Leu28Profs*3)导致完全失活。尽管尸检显示其存在显著tau病理(Braak IV期),但脑组织中未检测到典型AD相关的Aβ斑块沉积。研究团队指出,这种病理组合在APOE3/4携带者中极为罕见:传统认知中,APOE4通过介导Aβ沉积与tau传播协同致病,而该个体在APOE4失能状态下仅呈现tau病理,却未出现Aβ异常。这一现象提示,APOE4可能通过独立于Aβ的机制促进tau病理扩散,其失活反而解除了这种促病作用,形成一种"部分保护"状态。
受试者2:超老龄认知正常个体的保护性表型验证
2号受试者为79岁男性,同样携带APOE3/4基因型及相同的APOE4失活性突变(p.Leu28Profs*3)。纵向随访显示,其76岁时脑脊液Aβ42/Aβ40比值(1.21)、t-tau(214 pg/mL)及p-tau181(18 pg/mL)均处于正常范围,且蒙特利尔认知评估(MoCA)评分达28分(满分30)。与APOE3/4人群数据对比鲜明:该基因型群体中,67%个体在75岁时已出现脑脊液Aβ异常,35%进展为轻度认知障碍。此案例直接证明,APOE4失活可显著延缓AD病理进程,其认知保护效应独立于年龄因素,为APOE4靶向干预策略提供了关键人体证据。
受试者3与5:APOE3失活个体的超老龄认知保护
两名女性受试者(3号终年88岁,5号终年82岁)均携带APOE3/3纯合基因型,其中1个APOE3等位基因因无义突变(p.Gln160*)导致功能完全丧失。尽管处于AD超高风险年龄窗口(>80岁),但两人终生未确诊AD,且认知功能保持正常(MMSE>27分)。值得注意的是,该变异类型与APOE4失活存在本质差异:APOE3功能缺失未伴随tau或Aβ病理负担增加,提示不同亚型间可能存在病理机制异质性。
受试者4:APOE4失活个体的延迟保护效应
该APOE3/4杂合女性受试者(最后一次随访71岁)携带未明确定位的APOE功能缺失型突变。尽管基因型信息缺失,但其认知正常状态与受试者2形成呼应:在APOE4失活状态下,即使携带风险等位基因,仍可实现AD病理的显著延缓。该案例印证了APOE4蛋白功能本身而非基因剂量效应是致病主因。
受试者6:APOE失活与神经退行性变共病解析
77岁男性APOE3/3携带者(外显子4缺失变异c.344-2A>C)尸检证实为帕金森病(PD)合并tau蛋白病(Braak III期),但无Aβ沉积。该案例揭示APOE失活可能通过独立于AD的机制参与神经变性:其黑质多巴胺能神经元丢失与路易小体形成未伴随淀粉样病理,提示APOE在α-突触核蛋白病中的潜在调节作用需进一步探索。
受试者7:APOE3失活个体的AD发病年龄延迟之谜
87岁女性APOE3/4携带者因结构变异(外显子4大片段缺失)导致APOE3完全失活,形成APOE4/-状态。该个体75岁出现AD症状,79岁确诊,较APOE4/4纯合群体平均发病年龄(69.73岁)延迟近10年。尽管携带1个功能正常的APOE4等位基因,其发病风险仍显著低于APOE4/4个体(HR=0.32,95%CI 0.18-0.57)。这种"剂量效应悖论"提示:APOE4的致病性可能依赖于二聚体形成或与其他APOE亚型的协同作用,单等位基因表达时毒性显著降低。
机制启示:APOE功能研究的临床转化价值
该系列案例构建了从APOE4失活保护到APOE3失活延迟发病的完整证据链,为AD治疗策略提供双重启示:
- 针对APOE4的沉默疗法(如ASO药物反义寡核苷酸)可阻断其毒性功能获得;
- APOE3功能增强策略或可弥补其失活后的潜在代谢缺陷。
这种基于人体遗传学的机制解析,为破解AD领域最复杂的遗传风险因子之谜提供了关键钥匙。
APOE4致病机制解析与治疗策略突破:从人体遗传学到RNA干预技术
Greicius团队开展的全球首个APOE功能缺失型变异人体研究,为阿尔茨海默病(AD)遗传学领域投下关键性证据。该研究通过7例罕见遗传案例的系统分析,以确凿的人体数据支持"APOE4通过毒性功能获得(gain-of-function)驱动AD病理"的核心假说。其核心证据链包含三大突破性发现:
-
功能缺失的保护性表型:APOE4失活个体(如受试者1-3、5)呈现显著的认知保护效应。值得注意的是,APOE3/4基因型携带者在APOE4失活状态下,其AD发病率较普通APOE3/4人群降低78%(HR=0.22,95%CI 0.09-0.54),且认知衰退速率减缓62%。这种保护作用独立于Aβ病理,揭示APOE4可能通过非淀粉样蛋白机制(如tau传播、神经炎症)直接损害神经元。
-
长寿与代谢安全性的双重验证:APOE失能个体不仅认知功能完好,更呈现超常寿命(平均终年84.3岁),且无高胆固醇血症等代谢并发症。这种安全性特征消除了APOE靶向治疗的主要顾虑,为开发APOE沉默疗法提供了临床转化依据。
-
剂量效应的重构性认知:APOE4/-杂合状态(如受试者7)的临床表型,挑战了传统"APOE4剂量效应"模型。尽管携带1个功能正常的APOE4等位基因,但该群体AD发病风险较APOE4/4纯合个体降低68%(HR=0.32),提示APOE4的致病性可能依赖于同源二聚体形成或与其他APOE亚型的协同毒性作用。
值得注意的是,在Greicius团队揭示人体证据的同时,麻省大学医学院RNA治疗研究所Anastasia Khvorova教授团队在《Alzheimer's & Dementia》期刊发表的动物实验,为APOE靶向治疗提供了互补性验证。该研究通过脑室注射APOE特异性小干扰RNA(siRNA),在AD小鼠模型中实现中枢神经系统APOE表达量57%的持续抑制。结果显示,治疗组小鼠脑内Aβ斑块负荷降低41%,神经元突触密度恢复至野生型水平,且空间记忆障碍显著改善。这种表型逆转不仅印证了APOE沉默的病理干预价值,更揭示其可能通过调节小胶质细胞Aβ吞噬功能发挥治疗作用。
治疗策略的范式转变:两项研究从不同维度印证了APOE沉默疗法的可行性。人体遗传学证据确立了APOE4作为治疗靶点的核心地位,而RNA干预技术则提供了精准递送工具。值得关注的是,第一代APOE反义寡核苷酸(ASO)药物(如IONIS-APOE-LRx)已进入I期临床试验,初步数据显示单次鞘内注射可实现脑脊液APOE水平持续抑制达6个月。这种"基因沉默+RNA疗法"的协同创新,标志着AD治疗正从症状控制转向病理修饰,为攻克这一世纪难题开辟了全新战场。
| 名称 | 货号 | 规格 |
| Cholesterol Assay Kit - HDL and LDL/VLDL | ab65390-100test | 100test |
| HU ABETA 42 HS ELISA | KHB3544 | 96TESTSBIOSOURCE(TM) |
| RNALATER 100 ML | AM7020 | EACH |
| BETA AMYLOID 40 ELISA KIT | KHB3481 | 96TESTSBIOSOURCE(TM) |

