





Immune evasion of dormant disseminated tumor cells is due to their scarcity and can be overcome by T cell immunotherapies
尽管抗肿瘤免疫应答被激活,休眠的播散性肿瘤细胞(DTCs)仍能长期存活。其逃逸免疫监视的具体机制尚未明确。本文介绍2025年1月发表于Cancer Cell Metabolism(IF=50.3)的研究,该研究揭示了一种未被充分认识的免疫逃逸新范式——"相对稀缺性"效应,其构成DTCs持续存在的病理基础。值得注意的是,基于T细胞的免疫疗法可能通过增强DTCs与抗原特异性T细胞的相互作用频率,反而触发DTCs的适应性耗竭机制。这一发现为理解肿瘤免疫微环境中的非线性相互作用提供了新视角,并为优化现有免疫治疗策略提供了理论依据。

一、Highlight:
- 首次揭示"相对稀缺性"现象是播散性肿瘤细胞(DTC)逃逸免疫监视的核心机制;
- DTC与抗原特异性T细胞的相互作用频率存在临界阈值,低频接触不足以触发清除效应;
- 通过提升T细胞-DTC相互作用频率可突破免疫耐受,实现DTC的免疫根除;
- 疫苗接种或过继性T细胞疗法可作为潜在干预策略,为清除微小残留病灶提供新范式。
二、研究背景
流行病学证据表明,肿瘤晚期复发主要源于原发肿瘤早期向肺、骨髓等外周组织播散的肿瘤细胞。在乳腺癌临床前期阶段,约30%患者的骨髓穿刺样本中可检出单个播散性肿瘤细胞(DTC),其中多数细胞呈Ki67增殖标记阴性表型。临床前研究已证实,清除DTC与无转移生存率显著相关:通过干预手段减少或消除DTC可显著改善小鼠模型及乳腺癌患者的预后。然而,靶向DTC的免疫治疗策略受限于其特异性标记物的鉴定困难,临床转化进展缓慢。
当前,DTC与宿主免疫系统的相互作用机制尚未完全阐明。部分研究推测,DTC可能通过模拟组织干细胞的慢循环特性逃避免疫监视,例如下调主要组织相容性复合体I类分子(MHC I)表达——这是CD8+ T细胞识别并杀伤靶细胞的关键分子。据此,有学者提出恢复DTC的MHC I表达可能增强免疫系统对其的监控能力。然而,临床观察数据却显示T细胞具备识别DTC的能力,这使得MHC I下调是否构成CD8+ T细胞识别DTC的核心障碍仍存争议。
嵌合抗原受体(CAR)T细胞疗法为DTC靶向治疗提供了新思路。与依赖MHC I的天然T细胞不同,CAR-T细胞通过识别细胞表面抗原直接杀伤靶细胞,在血液系统恶性肿瘤治疗中已取得突破性进展。值得注意的是,临床试验证实,针对肿瘤相关抗原的CAR-T细胞疗法可有效清除骨髓中的微小残留病灶。但在实体肿瘤领域,由于免疫抑制性肿瘤微环境的存在,该疗法尚未取得同等疗效。这种疗效差异是否同样适用于DTC等微小残留病灶尚不明确。因此,深入解析DTC逃逸抗原特异性T细胞免疫监视的分子机制,对开发新型免疫治疗策略具有重要科学价值。
三、主要研究结果
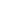
1. 功能性抗原特异性T细胞存在下休眠DTC的持续存在
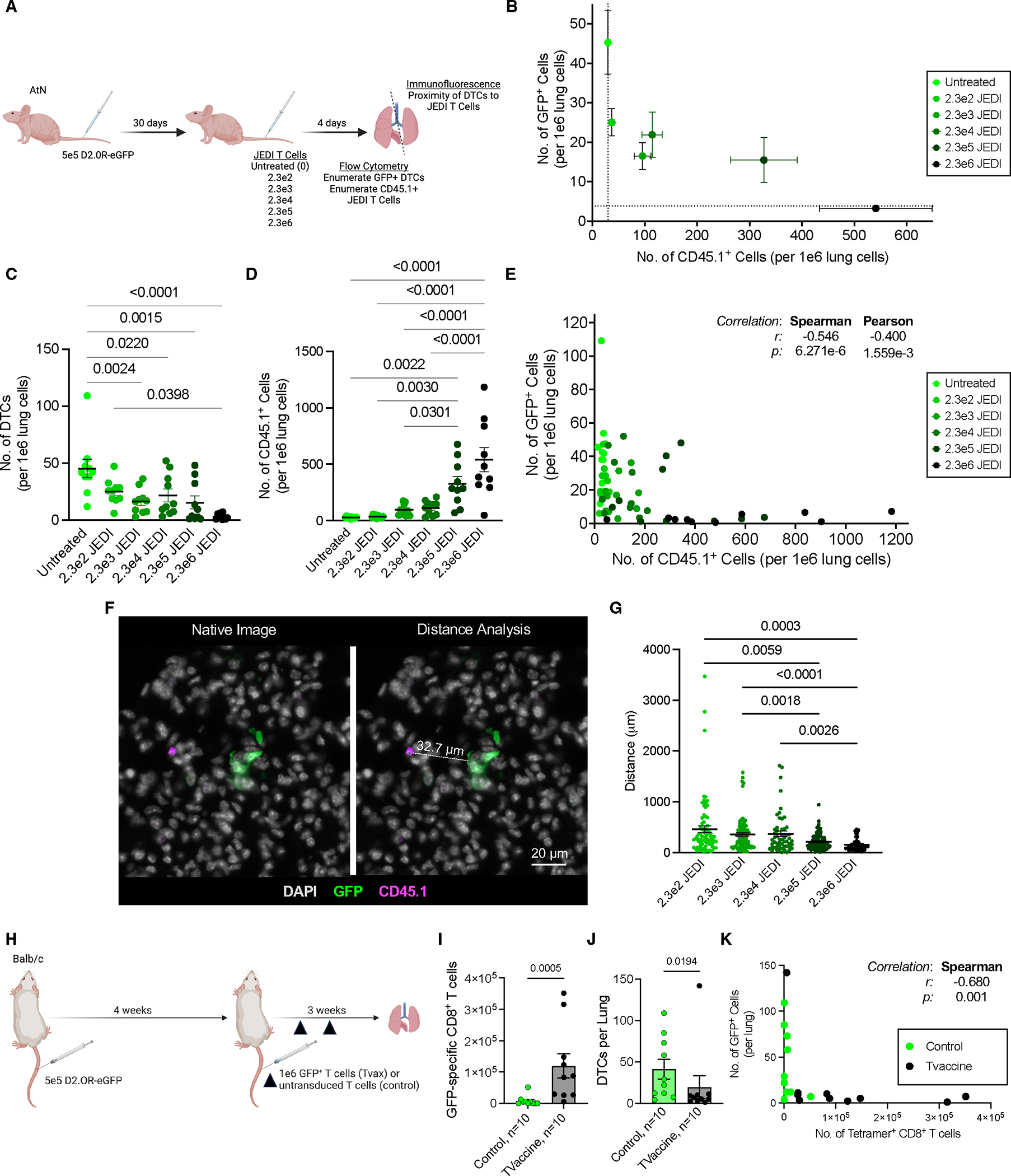
研究证实,即便原发肿瘤实现病理完全缓解,免疫检查点抑制剂仍无法有效激发过继性免疫应答以清除残留DTC。为明确DTC免疫监视的障碍机制,研究者首先验证表达优势新抗原的DTC能否被内源性肿瘤抗原特异性T细胞识别清除。实验采用亲本/野生型或ffluc/eGFP+同种异体乳腺肿瘤细胞接种Balb/c小鼠,待其完全排斥原位肿瘤后(图1A-B),于第50天及第100天采集骨髓样本,通过共聚焦成像分析发现:尽管在脾脏、淋巴结及骨髓中均检测到eGFP特异性CD8+ T细胞(MHC I四聚体染色证实,图1C),但骨髓中仍持续存在eGFP+ DTCs。值得注意的是,部分DTC与CD8+ T细胞存在空间邻近,但极少表现为eGFP特异性T细胞(图1D)。
进一步表型分析显示,肿瘤排斥后50天,四聚体+抗原特异性T细胞呈现效应记忆(CD44hiCD62Llo)与中枢记忆(CD44hiCD62Lhi)双重表型(图1E)。尽管eGFP特异性记忆T细胞高表达PD-1(水平与四聚体阴性T细胞相当),但缺乏终末耗竭标志物TIM-3(图1F-G),提示其保留局部及系统性肿瘤监视潜能。生物发光成像(BLI)证实,既往携带并排斥4TO7-ffluc/eGFP肿瘤的小鼠可完全抵御同源肿瘤细胞攻击(图1H-I),且肿瘤再挑战后骨髓中eGFP特异性CD8+ T细胞频率显著升高(图1J-K),进一步验证了记忆T细胞库的功能活性。然而,尽管抗原特异性CD8+ T细胞数量增加3倍(图1K),骨髓中eGFP+ DTC数量却未发生显著变化(图1L)。
上述数据表明,功能性抗原特异性CD8+ T细胞的存在不足以清除骨髓中的残留DTC,揭示了肿瘤免疫监视过程中存在未被认知的耐受机制。
2. DTCs通过下调MHC I实现免疫逃逸
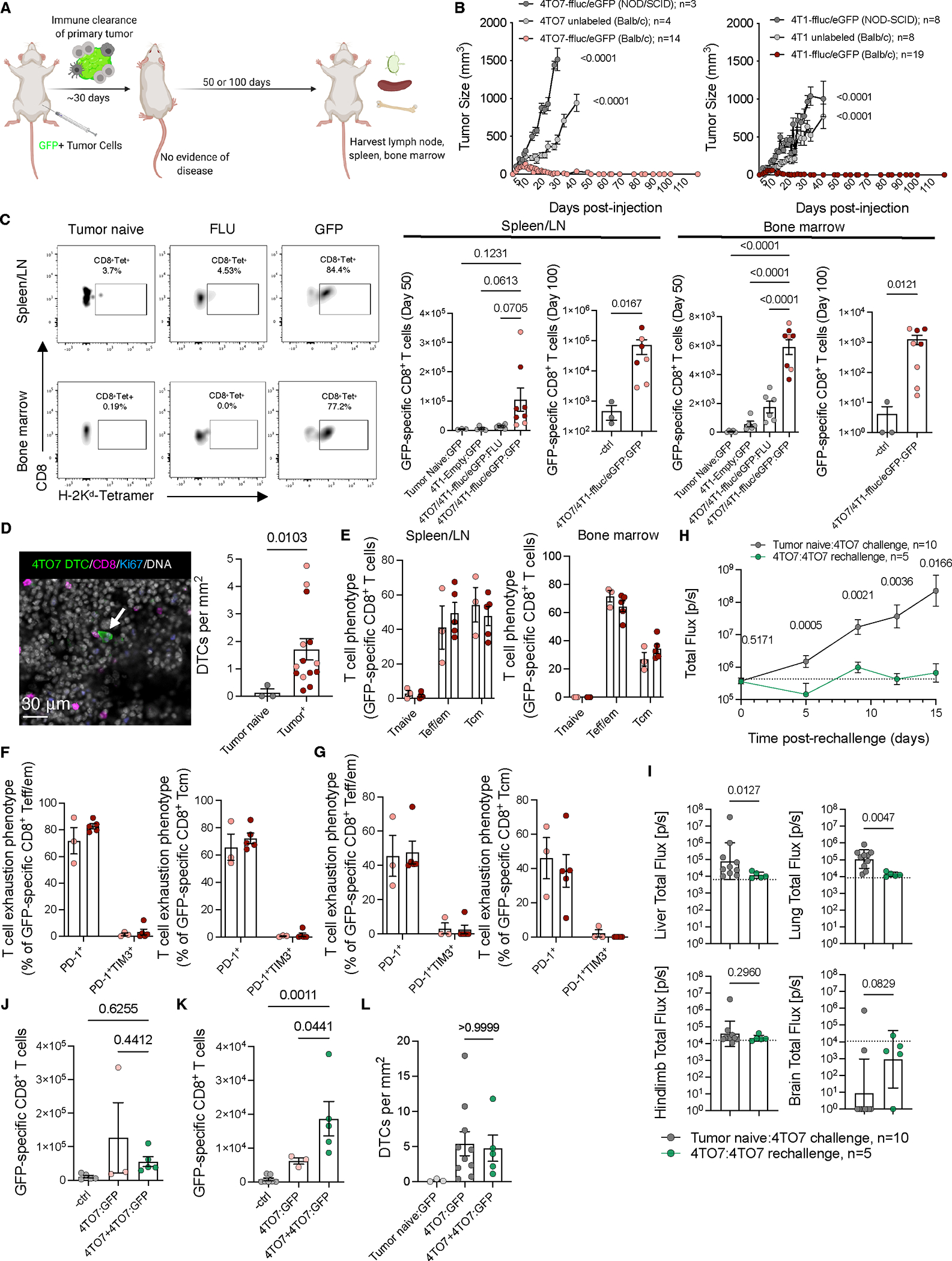
实验采用静脉注射D2.0R-eGFP乳腺肿瘤细胞构建同源Balb/c肺转移模型(图2A)。在无创伤或炎症刺激条件下,D2.0R细胞以单个或微小细胞簇形式长期(4周至240天)驻留于肺部,且绝大多数呈Ki-67阴性休眠状态。尽管在脾脏/淋巴结中可检出数百个eGFP特异性CD8+ T细胞(图2B),肺部组织中更存在数千个抗原特异性T细胞(图2C),但DTCs数量仍持续稳定存在(图2D),提示其可能通过MHC I下调、免疫抑制因子分泌或检查点分子过表达等机制逃逸免疫监视。
为解析具体逃逸机制,研究者于静脉注射后4周分离Balb/c小鼠及T细胞缺陷型裸鼠(AtN)肺部的D2.0R DTCs/微转移灶及D2A1转移灶,进行低输入RNA测序分析(图2E)。组织学检查显示,D2.0R细胞主要以Ki-67阴性单细胞或微小病灶形式存在,而D2A1细胞则形成增殖性转移灶(图2F)。转录组分析表明,与D2A1相比,D2.0R细胞未表现出抗原加工递呈、免疫抑制因子或检查点分子相关基因集的显著差异表达,但缺氧相关基因表达显著降低(图2G),提示休眠DTCs可能通过独特机制规避免疫识别。
进一步验证发现,D2.0R细胞随时间推移持续下调MHC I分子(H-2Kd),而D2A1细胞转移过程中MHC I表达逐步上调(图2H)。免疫荧光染色证实,休眠期Ki-67- DTCs的H-2Kd表达水平仅为增殖期Ki-67+病灶的22.2%(图2I-J)。值得注意的是,在多种转移模型中,MHC I下调现象独立于细胞增殖状态(Ki-67+/- D2A1细胞MHC I表达无差异),且在T细胞缺陷裸鼠中仍持续存在(图2H-J),表明MHC I表达调控不依赖于适应性免疫压力或细胞周期活动。
综上,研究证实DTCs通过主动下调MHC I表达实现免疫逃逸,此过程既非单纯由细胞周期停滞驱动,亦非适应性免疫压力诱导的结果,而是构成休眠肿瘤细胞长期存续的核心机制。
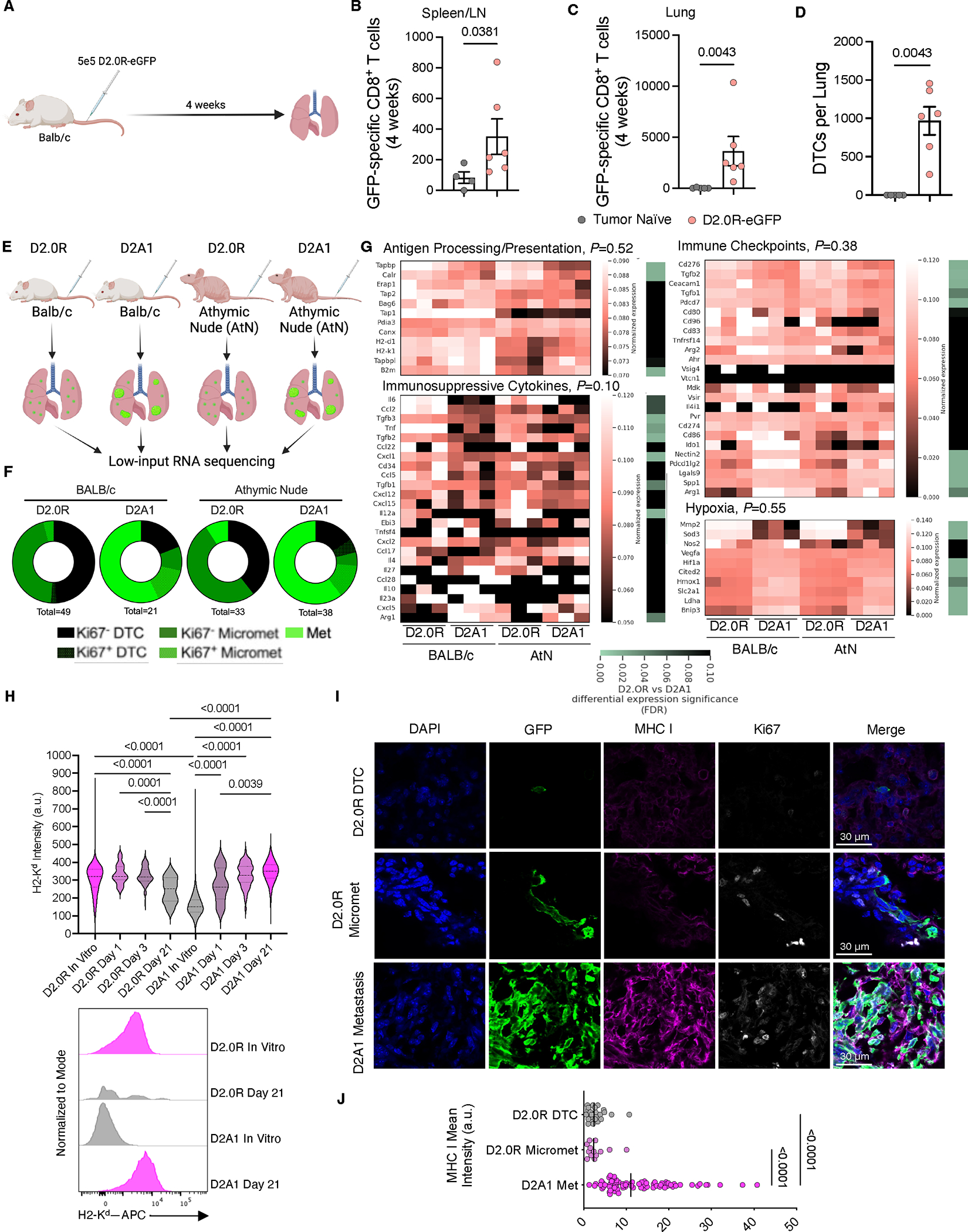
3. MHC I限制性肿瘤特异性T细胞可有效清除休眠DTCs
研究采用微血管龛(MVN)三维培养体系模拟体内休眠微环境,通过血管内皮细胞与骨髓基质细胞共培养诱导人乳腺癌细胞(T4-2)进入静止状态(图3A-B)。Ki-67染色显示,相较于骨髓基质单层培养中MHC I(HLA-A2)表达下调33%的静止期T4-2细胞,MVN培养体系中静止细胞MHC I表达进一步降低39%(图3B-D),重现了小鼠体内休眠DTCs的MHC I下调特征(图2H-J)。
为验证T细胞杀伤效能,研究者构建表达纽约食管鳞状细胞癌抗原-1(NY-ESO-1)的T4-2细胞模型。添加HLA-A2/NY-ESO-1特异性TCR-T细胞可特异性杀伤84%-97%的NY-ESO-1+ T4-2细胞,其效果显著优于未处理组及NY-ESO-1阴性对照组(图3E-G)。值得注意的是,TCR-T细胞的杀伤效能不受肿瘤细胞所处微环境影响:在间充质干细胞支持的增殖性肿瘤细胞群及MVN诱导的静止态肿瘤细胞中,均观察到高效的抗原特异性清除作用(图3F-G)。
相较于体外培养系统(MHC I下调幅度有限,图3C),体内休眠DTC的MHC I表达水平更低(图2J)。研究者进一步采用合成D2.0R小鼠乳腺癌细胞构建肺转移模型,该细胞表达eGFP及流感病毒血凝素(HA)作为MHC I限制性肿瘤新抗原(图3H)。结果显示,CL4 TCR-T细胞治疗可显著清除肺部D2.0R DTCs,其疗效显著优于多克隆T细胞治疗及单纯淋巴清除化疗(图3I)。尽管低剂量IFNγ可上调DTCs的MHC I表达(体内外实验均证实,图3J),但未增强内源性免疫应答(图3K)或过继转移TCR-T细胞的杀伤效能(图3L-N)。综合两项独立实验数据,TCR-T细胞治疗组较对照组实现显著DTC清除(图3L-N),但残留DTCs仍维持正常MHC I表达水平(图3O),提示存在MHC I非依赖性保护机制。
上述结果表明,尽管休眠DTCs存在MHC I下调现象,但其下调幅度不足以完全规避肿瘤抗原特异性TCR-T细胞的识别与杀伤,揭示靶向MHC I通路的免疫治疗在清除微小残留病灶中的潜在价值。
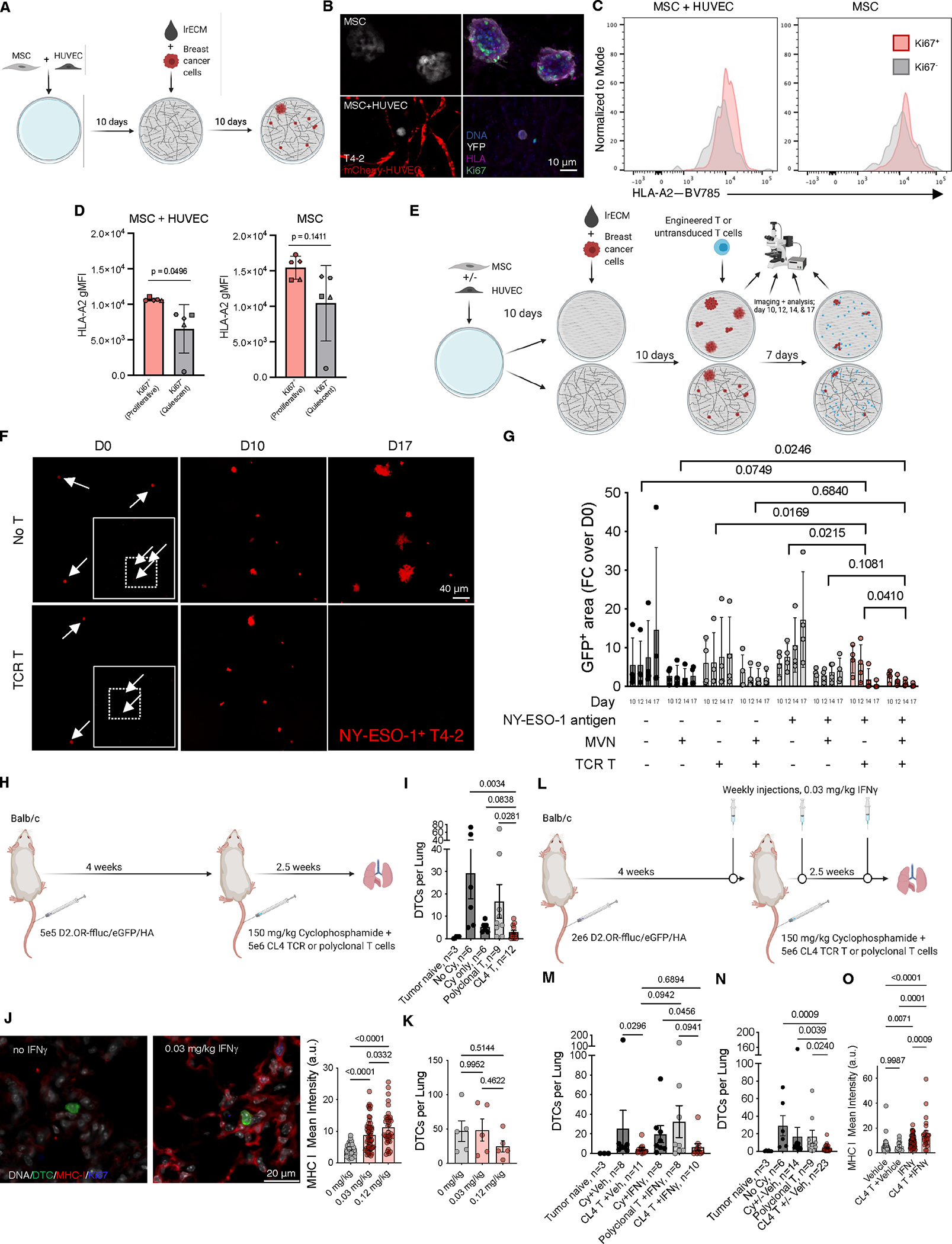
4. 相对稀缺性是DTCs逃避抗原特异性T细胞免疫监视的核心机制
为验证"相对稀缺性"假说,研究向T细胞缺陷型裸鼠(AtN)静脉注射D2.0R-eGFP细胞构建肺转移模型,30天后通过过继回输不同剂量JEDI TCR-T细胞(CD45.1+标记)评估T细胞-DTC相互作用频率对清除效能的影响(图4A)。流式细胞术证实,回输JEDI T细胞在肺部的定植效率与输入剂量呈正相关(图4B-C)。随着JEDI T细胞数量增加,肺部残留eGFP+ DTCs数量呈剂量依赖性减少(图4B-D),二者呈显著负相关(r²=0.89,p<0.0001,图4E)。空间定位分析显示,残余DTCs与最近JEDI T细胞的平均距离随T细胞剂量增加而缩短(图4F-G),表明T细胞密度提升可增强局部接触频率,从而突破免疫逃逸阈值。
进一步采用肿瘤疫苗(Tvaccine)验证内源性T细胞扩增对DTC清除的影响:在D2.0R肿瘤接种后4周及15周,以Tvaccine或对照T细胞进行免疫增强(图4H)。结果显示,Tvaccine治疗使肺部eGFP特异性CD8+ T细胞数量扩增约18倍(图4I),同时实现53%的休眠DTC清除率(图4J),且T细胞数量与DTC负荷呈显著负相关(r²=0.76,p=0.002,图4K)。值得注意的是,单例治疗失败病例(图4I-K)显示其肺部eGFP特异性T细胞数量较同组其他个体低5-70倍,进一步佐证T细胞数量对清除效能的决定性作用。
研究证实:(1)DTCs与抗原特异性T细胞的相对稀缺性构成免疫逃逸基础,当T细胞密度突破临界阈值时,接触频率增加可逆转免疫耐受;(2)通过疫苗接种或过继性T细胞疗法提升效应T细胞数量,可显著增强DTC清除效率;(3)该机制为CAR-T细胞等基于T细胞的免疫疗法提供了理论依据,提示提高效应细胞/靶细胞比值可实现微小残留病灶的有效清除。
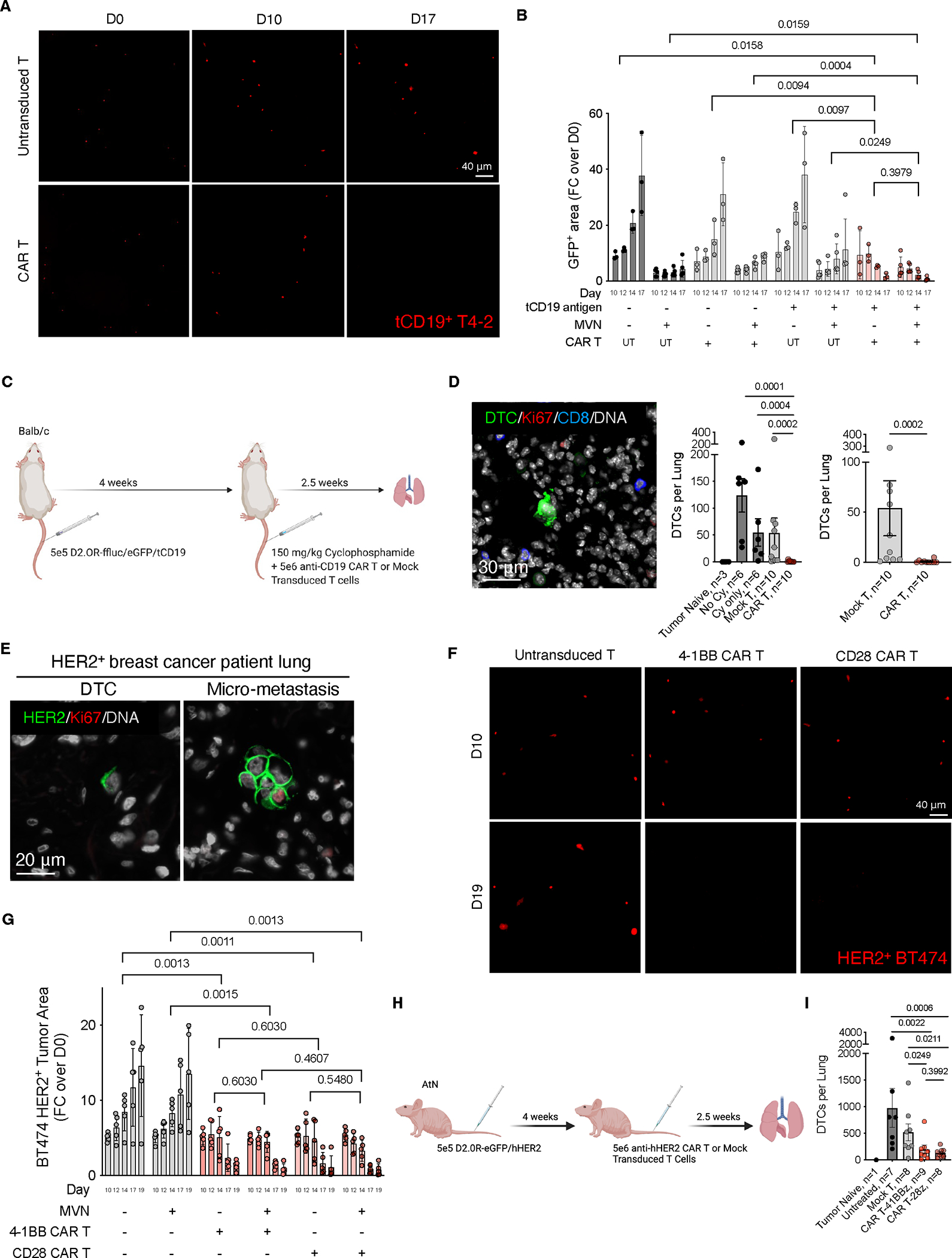
5. CAR T细胞通过突破"相对稀缺性"阈值实现DTCs高效清除
在三维培养体系中,针对模型抗原tCD19的CAR T细胞可清除93%的基质层或MVN微环境中增殖态/静止态T4-2乳腺癌细胞,而对照T细胞或非特异性CAR-T细胞无此效应(图5A-B)。进一步在D2.0R肺转移模型中验证,静脉注射tCD19+肿瘤细胞4周后,抗CD19 CAR-T细胞实现98%的休眠DTC清除率(10只小鼠平均,图5C-D),其疗效显著优于同源TCR-T细胞。
临床转化层面,研究者发现HER2+转移性乳腺癌患者肺部微小病灶中,DTCs及微转移灶持续表达HER2抗原(图5E)。在对应小鼠模型中,HER2特异性CAR-T细胞可清除89%-97%的BT474及HCC1569肿瘤病灶(图5F-G),且疗效不受共刺激结构域(4-1BB或CD28)差异影响(图5G)。在免疫缺陷小鼠的休眠DTC模型中,CAR-T细胞转移同样显著减少肺部HER2+ DTCs负荷(图5H-I),重现了体外实验结果。
研究通过模型抗原(tCD19)及临床相关抗原(HER2)双体系验证,证实CAR-T细胞对休眠DTC的清除效能至少等同于TCR-T细胞及肿瘤疫苗策略。机制解析表明,CAR-T细胞不依赖MHC I分子识别肿瘤细胞,通过直接提升效应细胞/靶细胞比值,突破"相对稀缺性"阈值,从而强制增强T细胞-DTC相互作用频率,最终实现免疫逃逸逆转。该发现为基于T细胞的肿瘤清除策略提供了关键理论依据,即通过工程化受体设计提升效应细胞靶向效能,可有效克服微小残留病灶的免疫耐受机制。
四、总结
本研究首次揭示了一种未被认知的免疫逃逸新范式——"相对稀缺性",其通过限制抗原特异性T细胞与播散性肿瘤细胞(DTC)的相互作用频率,构成肿瘤微小残留病灶长期存续的分子基础。研究证实,突破该免疫耐受阈值的关键在于提升效应T细胞密度:当T细胞数量超过临界比值时,接触频率增加可强制逆转DTC的免疫逃逸状态,从而实现功能性清除。
通过模型抗原(tCD19)及临床相关抗原(HER2)双体系验证,研究系统比较了三种T细胞免疫治疗策略的效能:(1)肿瘤疫苗通过内源性T细胞扩增提升效应细胞数量;(2)过继性TCR-T细胞转移直接补充抗原特异性效应细胞;(3)CAR-T细胞疗法凭借非MHC依赖性识别机制,显著增强效应细胞/靶细胞比值。三者均在不同程度上证实:效应细胞数量的增加与DTC清除率呈正相关,其疗效差异主要源于达到的效应-靶标比值而非作用机制本身。
这些发现具有双重转化价值:其一,为筛选高免疫原性DTC特异性抗原提供了理论依据,通过靶向此类抗原可最大化提升免疫治疗效能;其二,为清除微小残留病灶开辟了新策略,即通过工程化T细胞疗法或疫苗接种突破"相对稀缺性"限制,强制激活局部免疫应答。未来研究需进一步解析不同免疫治疗策略对免疫微环境的重塑机制,以推动个体化精准免疫治疗的发展。
相关产品推荐
| 病原/肿瘤 | 产品名称 | 抗原 | 序列 | MHC | 位置 | 货号 |
| EBV | HLA-A*0201/YLELLVWRL-PE Labelled Tetramer | EBV.LMP1 | YLELLVWRL | HLA-A*0201 | 125-133 | UA089001 |
| EBV | HLA-A*0201/YLQQNWTL-PE Labelled Tetramer | EBV.LMP1 | YLQQNWTL | HLA-A*0201 | 159-167 | UA089003 |
| EBV | H-2Db(b)/RAHY-NIVTF-PE Labelled Tetramer | HPV16.E7 | RAHYNIVTF | H-2Db | 49-57 | UA089002 |
| HPV | H-2K(b)/EVYDFA-FRQL-PE Labelled Tetramer | HPV16.E6 | EVYDFARDL | H-2Kb | 48-57 | UA089004 |
| HPV | HLA-A*0201/KLP-DLCTL-PE Labelled Tetramer | HPV18.E6 | KLPDCTL | HLA-A*0201 | 13-21 | UA089005 |
| HPV | HLA-A*0201/KLTNT-GLYQL-PE Labelled Tetramer | HPV18.E6 | KLTNTGLYNL | HLA-A*0201 | 92-101 | UA089006 |
| HPV | HLA-A*0201/TLODIVIHL-PE Labelled Tetramer | HPV18.E7 | TLODIVIHL | HLA-A*0201 | 7~15 | UA089007 |
| HPV | HLA-A*0201/QFLNTL-FV-PE Labelled Tetramer | HPV18.E7 | QFLNTLFSV | HLA-A*0201 | 88-97 | UA089008 |
| HPV | HLA-A*1101/GVNHQLPAR-PE Labelled Tetramer | HPV18.E7 | GVNHQLPAR | HLA-A*1101 | 43-52 | UA089009 |
| Influenza A Virus | H-2D(b)/ASNENMETM-PE Labelled Tetramer | Flu.NP | ASNENMETM | H-2Db | 366-374 | UA089010 |
| Influenza A Virus | H-2K(d)/TYQR-TRALY-PE Labelled Tetramer | Flu.NP | TYQRTRALY | H-2Kd | 147-155 | UA089011 |
| Influenza A Virus | H-2D(b)/ASNEN-MDTM-PE Labelled Tetramer | Flu.NP | ASNENMDTM | H-2Db | 366-374 | UA089012 |
| LCMV | H-2D(b)/KAVYNFATM-PE Labelled Tetramer | GP 33 | KAVYNFATM | H-2Db | 33-41 | UA089013 |
| LCMV | H-2D(b)/FQPGQGFVK-PE Labelled Tetramer | LCMV NP | FQPGQGFVK | H-2Db | 396-404 | UA089014 |
| Tumor-related | HLA-A*1101/VVGADGVK-PE Labelled Tetramer | KRAS | VVGADGVK | HLA-A*1101 | 7~16 | UA089015 |
| Tumor-related | HLA-A*1101/VVGAGVGK-PE Labelled Tetramer | KRAS | VVGAGVGK | HLA-A*1101 | 7~16 | UA089016 |
| Tumor-related | HLA-A*0201/KLVVGAGV-PE Labelled Tetramer | KRAS | KLVVGAGV | HLA-A*0201 | 5~14 | UA089017 |
| Tumor-related | HLA-A*0201/SLLMWITQC-PE Labelled Tetramer | NY-ESO1 | SLLMWITQC | HLA-A*0201 | 157-165 | UA089018 |
| Melanoma | HLA-A*0201/LMWITQCFL-PE Labelled Tetramer | NY-ESO2 | LMWITQCFL | HLA-A*0201 | 159-167 | UA089019 |
| Melanoma | H-2Db(b)/MMFPNA-P1-PE Labelled Tetramer | WT1 | RMFPNAPL | H-2Db | 126-134 | UA089020 |
| Melanoma | HLA-A*0201/CMTWV-PE Labelled Tetramer | WT2 | CMTWVNMDM | HLA-A*0201 | 235-243 | UA089021 |
| Melanoma | HLA-A*1101/KTCQRKSF-PE Labelled Tetramer | WT3 | KTCQRKSF | HLA-A*1101 | 386-394 | UA089022 |
| Ovarian Cancer | H-2K(b)/SINFEKL-PE Labelled Tetramer | OVA | SINFEKL | H-2Kb | 257-264 | UA089023 |


