





Nicotinamide riboside kinase 1 protects against diet and age-induced pancreatic β-cell failure
摘要
本研究揭示了烟酰胺核糖激酶1(NRK1)在肥胖与衰老背景下维持胰腺β细胞功能的关键作用。通过对全身性NRK1敲除(KO)和β细胞特异性敲除(BKO)小鼠模型进行高脂饮食(HFD)与衰老干预,发现NRK1缺失加剧葡萄糖不耐受和胰岛素分泌缺陷。值得注意的是,β细胞特异性NRK1缺失未重现表型,表明β细胞功能障碍源于肝脏、肾脏等外周器官NAD+代谢紊乱引发的系统性改变,而非β细胞自主性缺陷。关键机制包括:
- 循环DPP-IV升高:抑制GLP-1活性(图6H-I);
- 组织纤维化:胰腺、肝脏和肾脏纤维化加重(图5K-L,图6D-E);
- 线粒体功能受损:肝脏和肾脏复合物I呼吸能力下降(图6F-G)。 研究首次阐明NRK1依赖的NAD+合成通路在代谢应激下维持全身葡萄糖稳态的生理意义,为靶向NR代谢干预糖尿病提供了新视角。

引言:NAD+代谢紊乱与β细胞功能障碍的关联
代谢疾病与NAD+稳态的病理联系
胰岛素抵抗和2型糖尿病(T2DM)的发病率随肥胖与老龄化显著上升。研究表明,NAD+水平下降是代谢性疾病的共同特征:
- NAD+前体干预效果:补充烟酰胺核糖(NR)或烟酰胺单核苷酸(NMN)可改善胰岛素抵抗模型葡萄糖耐受性(参考文献4-7)。
- β细胞特异性机制:高糖或炎症因子刺激下,NMN能恢复β细胞葡萄糖刺激胰岛素分泌(GSIS)能力(参考文献8-9)。
NRK1的核心地位
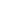
NRK1是NR转化为NAD+的限速酶(图1A)。其组织分布具有特异性:
- 高表达组织:肝脏、胰腺胰岛;
- 低表达组织:骨骼肌中由NRK2部分代偿。
本研究首次聚焦NRK1在β细胞应激适应中的作用,通过双模型(全身KO vs β细胞特异性BKO)揭示其系统性保护机制。
 图一
图一
研究方法与模型构建
动物模型设计
| 模型类型 | 遗传背景 | 干预方案 |
|---|---|---|
| 全身NRK1 KO | C57BL/6NTac纯合 | HFD(8周起始)或自然衰老(24月) |
| β细胞特异性BKO | C57BL/6NTac × C57BL/6J杂交 | 同全身KO |
| 注:BKO通过Ins1-Cre与Nmrk1 floxed小鼠杂交实现(图1C-D)。 |
核心实验方法
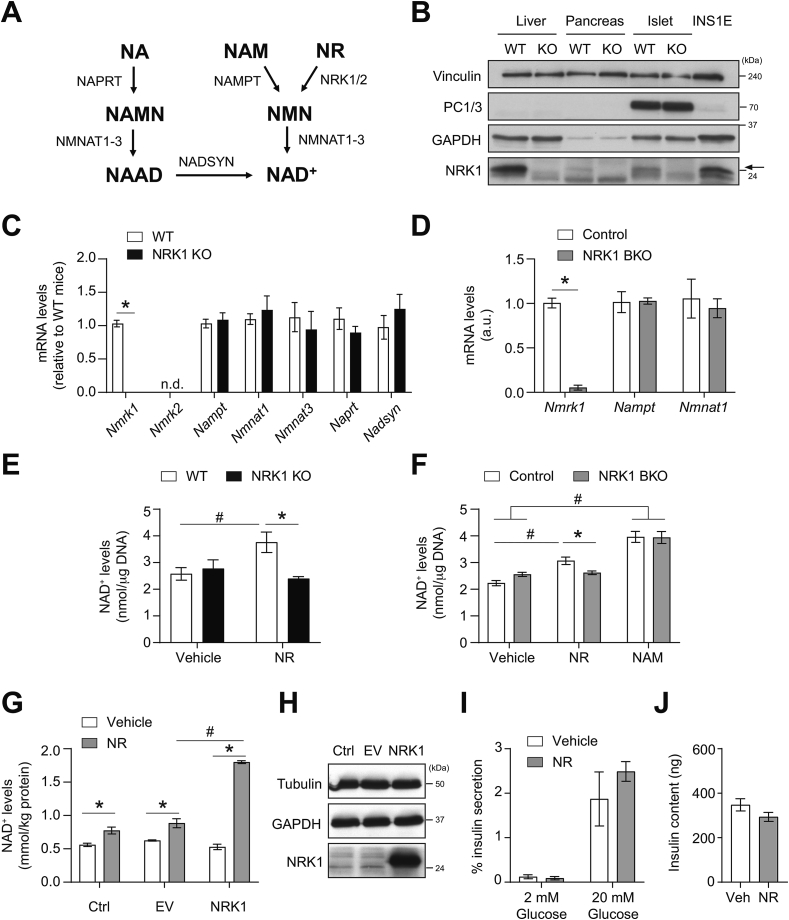
- 代谢表型分析:
- 葡萄糖/胰岛素耐量试验(IPGTT/IPITT)
- 间接热量测定(CLAMS系统监测VO₂、RER)
- 胰岛功能评估:
- 离体GSIS检测(图3I, 图5H)
- 胰岛素/胰高血糖素免疫组化(图3D, 图5A)
- 分子机制解析:
- NAD+水平检测(EnzyChrom试剂盒)
- RNA-Seq转录组分析(QuantSeq 3’ mRNA-Seq)
- 线粒体呼吸测定(Oroboros Oxygraph-2k)
 图二
图二
结果解析:NRK1缺失加剧代谢应激下的β细胞衰竭
1. NRK1在胰岛β细胞中功能性表达
- 表达验证:
NRK1蛋白在胰岛富集(较全胰腺高3倍),INS-1E细胞系中显著表达(图1B)。 - NAD+合成能力:
NR处理(0.5 mM, 2h)使野生型(WT)胰岛NAD+升高40%,而KO胰岛无响应(图1E)。过表达NRK1可放大NR效应(图1G-H),证实NRK1是β细胞利用NR的关键酶。
2. HFD诱导的β细胞功能障碍依赖全身NRK1
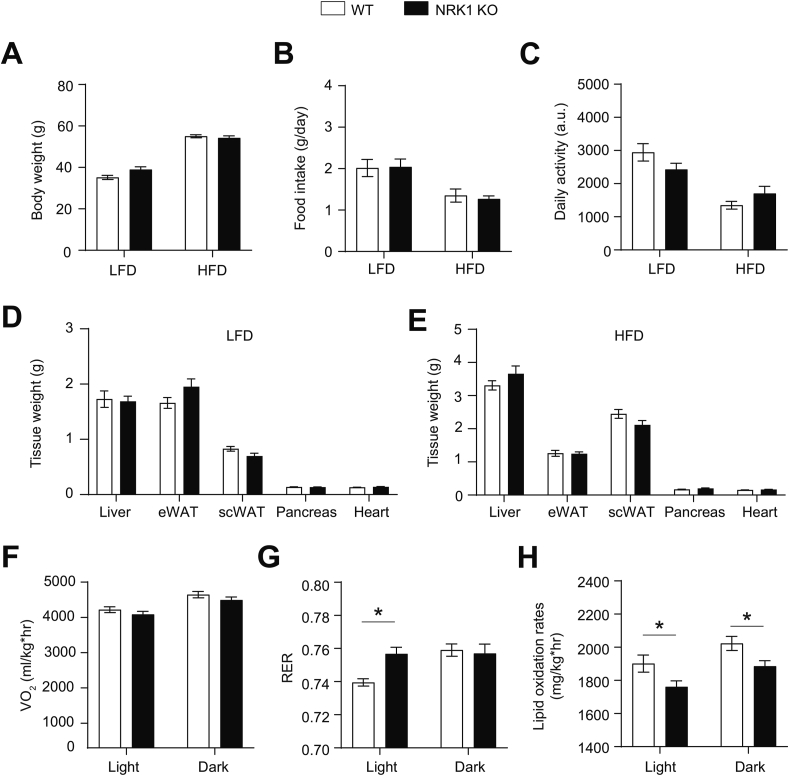
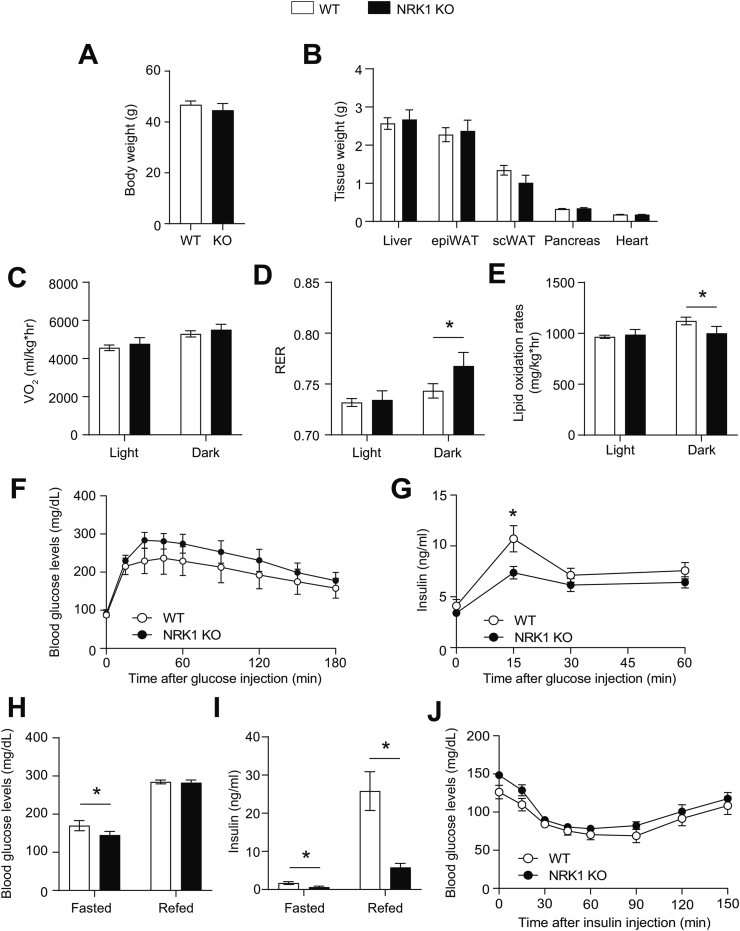
- 全身KO表型:
HFD喂养16周后,NRK1 KO小鼠出现:- 葡萄糖不耐受(血糖曲线下面积↑30%,图3A)
- GSIS受损(胰岛素分泌↓50%,图3B)
- 胰岛代偿性肥大(面积↑25%,图3F)但胰岛素含量↓(图3J)
- β细胞特异性BKO无表型:
血糖稳态、GSIS及胰岛基因表达(如MafA、Gck)均与对照无异(图S4G-L),提示β细胞自主性NRK1非必需。
 图三
图三
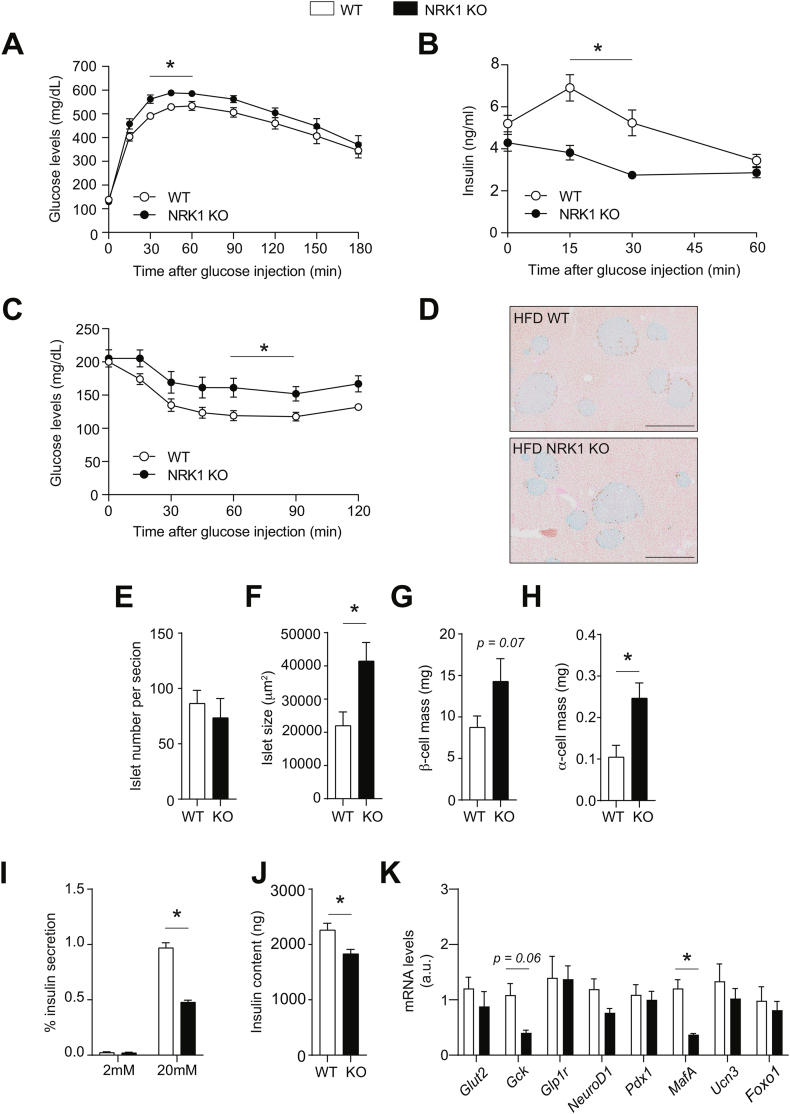
3. 衰老通过降低β细胞质量损害胰岛素分泌
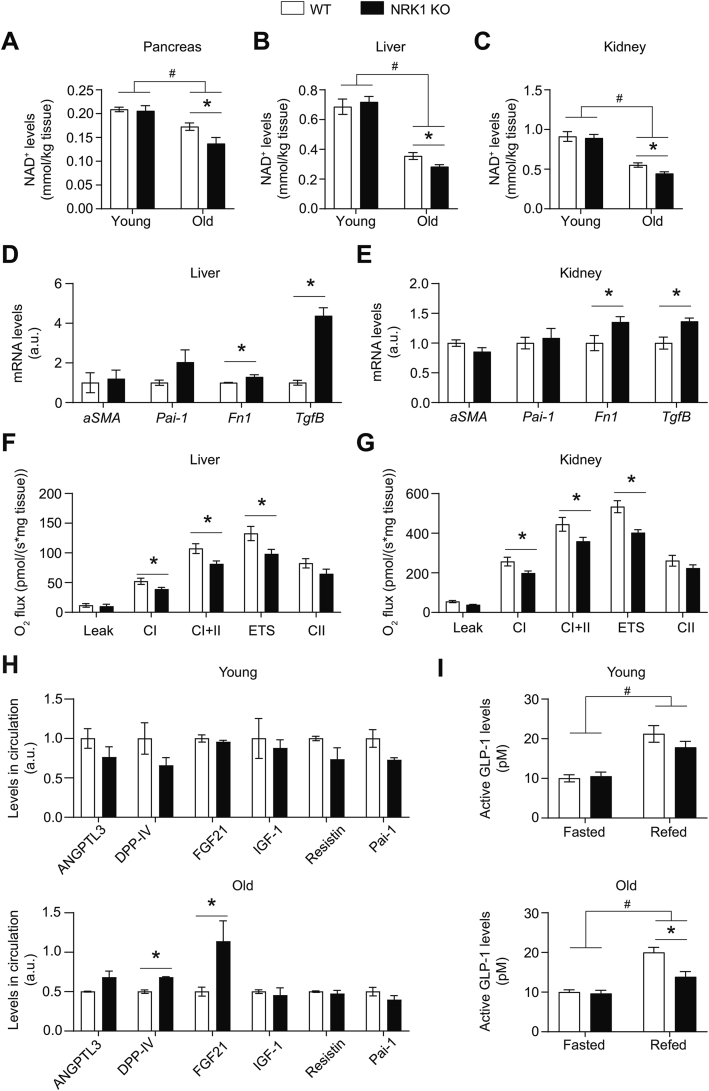
- 全身KO老龄鼠特征:
- 葡萄糖刺激后胰岛素分泌↓40%(图4G)
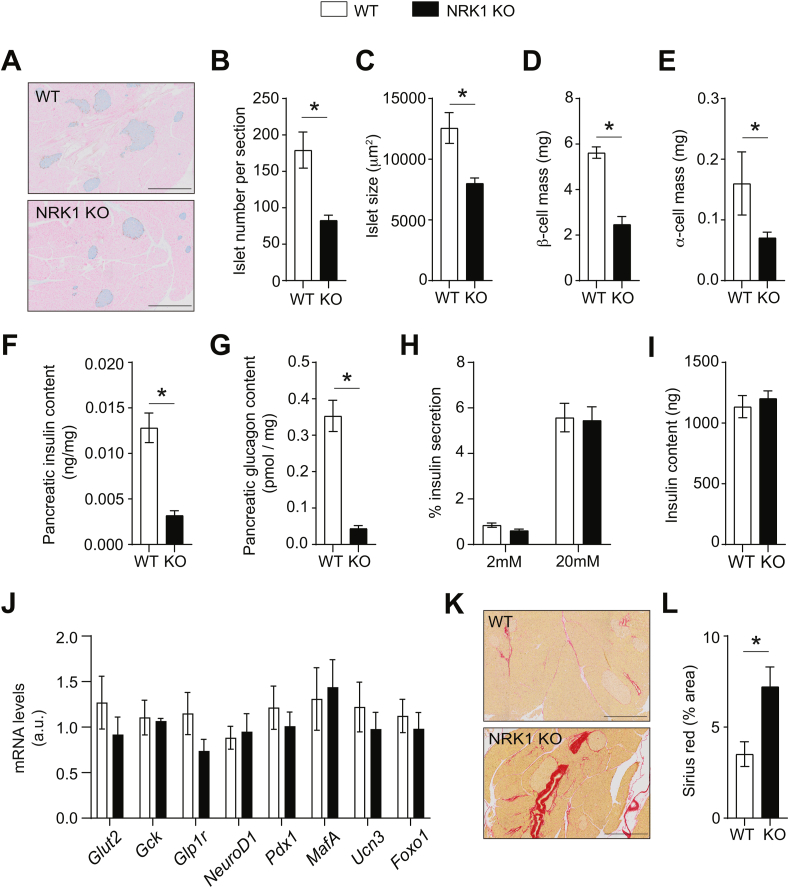
- 胰岛数量↓35%、纤维化面积↑3倍(图5B-C, L)
- 肝脏/肾脏NAD+水平较WT进一步降低20%(图6A-C)
- BKO无衰老相关缺陷:
胰岛素分泌、胰岛形态与对照无差异(图S5C-E),再证系统性机制主导。
 图四
图四
4. 全身性代谢紊乱的核心介质
- 循环因子异常:
- DPP-IV:HFD及老龄KO小鼠血浆水平均↑(图6H, S6B)
- GLP-1:老龄KO再进食后活性GLP-1↓50%(图6I)
- 器官纤维化与线粒体功能障碍:
- 肝/肾纤维化基因(Fn1, Tgfb)表达↑2倍(图6D-E)
- 肝脏复合物I最大呼吸能力↓30%(图6F)
 图六
图六
讨论:系统性NAD+失衡是β细胞衰竭的驱动因素
NRK1的生理独特性
本研究揭示NRK1在应激下的不可替代性:
- 基础状态:NAM仍可维持NAD+水平(图1F);
- 代谢应激:NRK1缺失导致外周器官(肝/肾)NAD+储备耗竭,引发循环因子紊乱(如DPP-IV↑),间接损害β细胞。
DPP-IV-GLP-1轴的关键作用
DPP-IV升高通过降解GLP-1削弱肠-胰岛轴功能:
- 直接证据:老龄KO再进食后GLP-1应答受损(图6I);
- 临床关联:DPP-IV抑制剂已是T2DM一线药物,本研究为其机制提供新佐证。
组织纤维化的恶性循环
胰腺纤维化减少功能性β细胞质量(图5K-L),而肝/肾纤维化进一步恶化全身代谢环境。NAD+前体干预可缓解纤维化(参考文献56-59),提示NRK1通路在此过程的保护作用。
结论与展望
本研究确立NRK1为肥胖和衰老背景下β细胞功能的系统性守护者:
- 机制创新:首次发现DPP-IV升高和GLP-1应答缺陷是NRK1缺失导致β细胞衰竭的关键介质;
- 临床意义:NR补充需解决胃肠降解问题(参考文献77-78),开发稳定NR前体或联合DPP-IV抑制剂可能是新方向;
- 局限性:BKO模型的混合遗传背景可能弱化表型,需在纯合背景中验证。
综上,靶向NRK1通路为代谢性疾病干预提供了新靶点,未来需深入探索器官间NAD+代谢的交互机制。
图表示例
表1. NRK1 KO与BKO模型在HFD和衰老下的表型对比
| 表型指标 | 全身NRK1 KO (HFD) | 全身NRK1 KO (衰老) | β细胞NRK1 BKO |
|---|---|---|---|
| 葡萄糖耐受 | ↓↓ | ↓ | ↔ |
| GSIS | ↓↓ | ↓ | ↔ |
| 胰岛数量/大小 | ↑ (肥大) | ↓ (萎缩) | ↔ |
| 循环DPP-IV | ↑ | ↑ | ↔ |
| 肝/肾NAD+水平 | ↓ | ↓↓ | ↔ |
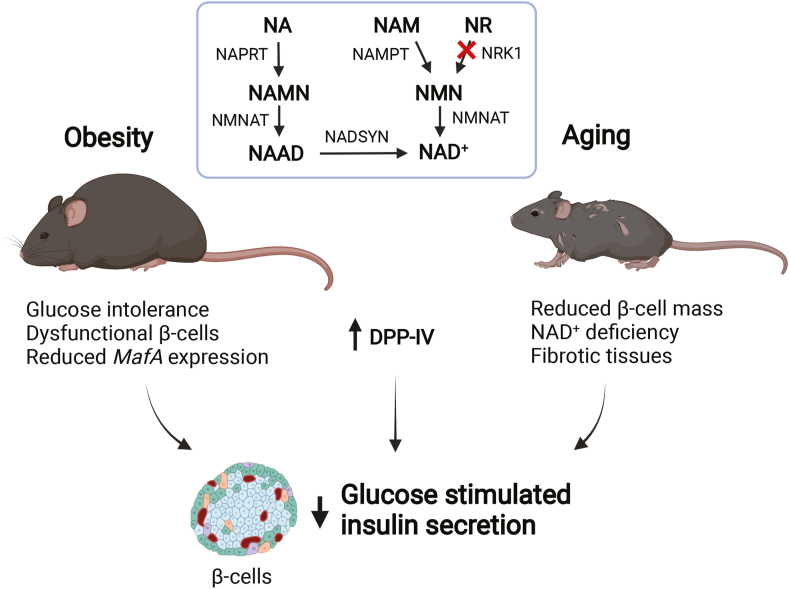
图7. NRK1缺失通过全身性代谢紊乱损害β细胞功能的机制总结 (示意图展示DPP-IV升高、GLP-1抑制、纤维化与线粒体功能障碍四重通路)
| 名称 | 货号 | 规格 |
| U-PLEX Metabolic 3-Plex Combo 2 (ms) SECTOR (25 PL) | K15305K-4 | 25PL |
| U-PLEX Metabolic 3-Plex Combo 2 (ms) SECTOR (5 PL) | K15305K-2 | 5PL |
| U-PLEX Metabolic 3-Plex Combo 2 (ms) SECTOR (1 PL) | K15305K-1 | 1PL |

