





Regulating microglial miR-155 transcriptional phenotype alleviates Alzheimer's-induced retinal vasculopathy by limiting Clec7a/Galectin-3+ neurodegenerative microglia
摘要 本研究首次在阿尔茨海默病(AD)模型小鼠(APPswe/PS1L166P)视网膜中发现Clec7a⁺和Galectin-3⁺神经退行性疾病相关小胶质细胞(MGnD/DAM)亚群的显著增加,并证实其与视网膜血管病变密切相关。研究采用他莫昔芬诱导的Cx3cr1CreERT2系统实现小胶质细胞特异性miR-155条件性敲除(cKO),发现该干预能显著降低视网膜Clec7a⁺和Galectin-3⁺ MGnD小胶质细胞数量,同时上调稳态标记P2ry12⁺小胶质细胞(图1f-g)。这种表型转换有效保护了血-视网膜内屏障(iBRB)完整性,减少血管淀粉样病变(图5a-h),并显著抑制视网膜炎症反应。蛋白质组学分析揭示,小胶质细胞特异性miR-155敲除能增强PI3K-Akt信号通路活性,并下调关键炎症因子IL-8和Spp1(骨桥蛋白)的表达(图4h-m)。该研究确立了靶向miR-155调控MGnD小胶质细胞表型作为治疗AD视网膜血管病变的新策略,为理解神经炎症与视网膜血管损伤的关联提供了重要机制见解。

引言:阿尔茨海默病视网膜病变与小胶质细胞表型调控
视网膜作为中枢神经系统(CNS)的延伸部分,因其具备非侵入性高分辨率成像的优势,已成为研究AD病理的重要窗口。大量研究证实,AD患者及模型动物的视网膜中存在AD特异性病理改变,包括β-淀粉样蛋白(Aβ)沉积、神经退行性变、神经节细胞丢失以及显著的血管病变。近年研究发现,AD患者视网膜中伴随Aβ沉积出现反应性星形胶质细胞和小胶质细胞的活化。然而,视网膜中是否存在与脑部类似的神经退行性疾病相关小胶质细胞(MGnD/DAM)表型,以及其具体调控机制和病理作用,此前尚未明确。
小胶质细胞是CNS的常驻免疫细胞,在维持稳态、清除病理蛋白和介导炎症反应中扮演核心角色。在AD等神经退行性疾病中,小胶质细胞会经历从稳态(M0)向激活状态的表型转换。MGnD/DAM表型是近年通过单细胞RNA测序鉴定出的一类与神经退行性疾病(特别是AD)密切相关的活化小胶质细胞亚群,其活化依赖于TREM2-APOE信号通路,并高表达Clec7a(Dectin-1)、ApoE、Galectin-3等分子,常聚集在Aβ斑块周围。microRNA-155 (miR-155) 被识别为该通路的关键转录调节因子,它通过靶向RICTOR(mTORC2复合物关键组分)和SMAD1(TGF-β信号通路效应分子)等,调控包括PI3K-Akt、血管重塑和血脑屏障(BBB)完整性在内的多条通路。在肌萎缩侧索硬化症(ALS)的SOD1-G93A小鼠模型中,靶向抑制miR-155成功恢复了具有神经保护作用的M0稳态小胶质细胞群,并增强了其吞噬能力。本研究首次聚焦于miR-155在AD视网膜中MGnD小胶质细胞表型转换中的作用,并探究其对视网膜血管病变的影响。
材料与方法
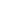
1. 基因工程小鼠模型构建 本研究采用复杂的三系杂交策略构建小胶质细胞特异性miR-155条件敲除(cKO)的AD模型小鼠(图1a):
- APPswe/PS1L166P小鼠:快速Aβ沉积的常用AD转基因模型。
- Cx3cr1CreERT2小鼠:表达他莫昔芬(Tamoxifen)诱导型Cre重组酶,该酶主要在小胶质细胞和部分单核/巨噬细胞中表达。
- miR-155fl/fl小鼠:携带两侧带有loxP位点的miR-155条件性敲除等位基因。 通过系统交配,最终获得四组核心实验动物:APP/PS1 (AD模型)、APP/PS1:miR-155cKO (AD模型+小胶质细胞miR-155敲除)、WT (野生型对照)、WT:miR-155cKO (野生型+小胶质细胞miR-155敲除)。所有小鼠在6周龄时通过腹腔注射他莫昔芬(150 mg/kg,共2次)诱导Cre重组酶活性,实现小胶质细胞特异性的miR-155基因敲除。动物实验均遵循相关伦理委员会批准。
2. 多维度视网膜分析技术 研究运用了多种先进技术对小鼠视网膜进行全方位解析:
- 细胞分选与qRT-PCR:
- 采用Percoll梯度离心法分离脑单核细胞。
- 利用流式细胞术(FACS)分选Ly-6C⁻CD11b⁺Fcrls⁺(或类似标记组合)的小胶质细胞群体。
- 使用TaqMan探针进行定量RT-PCR检测miR-155表达水平(Mu-miR-155, Assay ID:002571),以U6 snRNA作为内参。
- 免疫荧光染色与定量:
- 视网膜铺片技术:用于全视网膜观察,进行Clec7a/TMEM119/lectin(标记血管)、Galectin-3/TMEM119/lectin等多重免疫荧光染色(图1b-e, 图2a-e)。
- 视网膜切片:用于层特异性分析,进行Galectin-3/Iba1/12F4(标记Aβ42)、Tmem119/12F4、P2ry12/12F4、Apoe/Iba1/12F4等共标染色(图2g-h, i-j)。
- 立体学计数与图像分析:使用ImageJ软件对免疫反应阳性细胞进行手动计数或对免疫反应阳性面积进行定量分析(图1f-g, 图2g, 图5b, e, f, h)。对全视网膜或特定层(GCL, IPL, OPL)进行系统采样和分析。
- 蛋白质组学分析:
- 采用串联质谱标签(TMT)10-plex试剂进行蛋白质标记定量。
- 使用高pH反相色谱(HPRP)对肽段进行分级(合并为8个组分)。
- 在Orbitrap Lumos质谱仪上进行检测(分辨率设置为60,000)。
- 利用IPA(Ingenuity Pathway Analysis)软件进行通路富集分析、上游调控因子预测和功能注释(图4b, h-m)。
- 视网膜血管分离技术:
- 采用弹性酶(Elastase, 40 U/mL, 37°C, 2小时)消化法分离视网膜血管网络。
- 对分离的血管进行免疫染色:如Claudin-1/4G8(标记总Aβ)/lectin、ZO-1/11A50-B10(标记Aβ40)/lectin等,以评估血管完整性、紧密连接蛋白表达及血管Aβ负荷(图5a, d)。
- 蛋白质印迹(Western Blot):检测全视网膜或视网膜血管裂解液中关键蛋白的表达水平,如P2ry12、磷酸化NF-κB p65、成熟IL-1β、TNF-α、Claudin-1、MMP-9、SPP1等,并进行光密度定量分析(图2l, 图3a-c, 图4j-l, n, 图5c, i)。
- Meso Scale Discovery (MSD) 多重细胞因子检测:定量分析视网膜裂解液中多种炎症因子(如IL-2, IL-5, IL-6, IL-10, IL-12, IFN-γ, TNF-α)的水平(图3d-i)。
- 统计学分析:主要采用GraphPad Prism软件。多组比较使用双向或三向方差分析(ANOVA)结合Tukey’s多重比较检验。两组比较使用非配对双尾Student t检验。相关性分析采用Pearson相关系数(r)。数据以平均值±标准误(SEM)表示,显著性水平设定为*p<0.05。
 图一
图一
 图二
图二
实验结果
1. miR-155敲除显著减少视网膜Clec7a⁺ MGnD小胶质细胞
- 空间分布特征:在8月龄APP/PS1小鼠视网膜中,Clec7a⁺/TMEM119⁻小胶质细胞主要富集于神经节细胞层(GCL)和内丛状层(IPL),并与视网膜血管(lectin⁺)紧密黏附(图1b箭头);而Clec7a⁺/TMEM119⁺细胞则主要位于外丛状层(OPL)(图1c)。在WT小鼠视网膜中未检测到Clec7a⁺小胶质细胞(图1d)。重要的是,在APP/PS1:miR-155cKO小鼠视网膜中,Clec7a⁺/TMEM119⁻细胞数量极少(图1e)。
- 定量分析:
- 对视网膜切片Iba1⁺/Clec7a⁺细胞进行计数显示,8月龄APP/PS1小鼠的Clec7a⁺小胶质细胞数量较同龄WT小鼠显著增加2.7倍(*p<0.01)。而miR-155cKO使APP/PS1小鼠的Clec7a⁺细胞数量降低至WT水平(图1f)。
- Clec7a⁺/Iba1⁺小胶质细胞比例在8月龄APP/PS1小鼠中显著升高(53% vs WT, *p<0.05),而cKO后该比例降低了44%(图1g)。
- 这些结果表明,AD模型小鼠视网膜中存在Clec7a⁺ MGnD小胶质细胞亚群,且主要分布在血管丰富的内层视网膜;小胶质细胞特异性敲除miR-155能有效抑制该病理亚群的扩增。
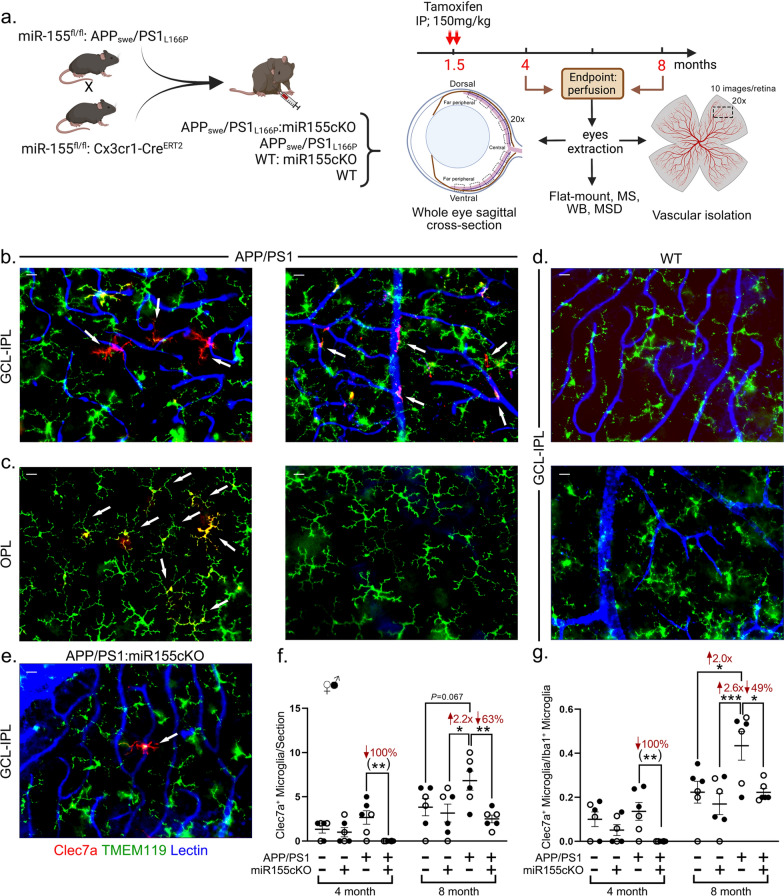
2. miR-155敲除下调视网膜Galectin-3⁺小胶质细胞并上调稳态标记
- 层特异性分布:Galectin-3⁺细胞同样主要集中于GCL-IPL区域(图2b),而OPL则以TMEM119⁺稳态小胶质细胞为主(图2c)。部分细胞呈现Galectin-3⁺/TMEM119⁺双阳性(图2e),提示存在表型过渡或异质性。部分Galectin-3⁺细胞被发现可吞噬视网膜Aβ₄₂斑块(图2h箭头)。
- 定量分析:
- 通过免疫荧光染色(Galectin-3/Iba1)和手动计数,发现APP/PS1小鼠视网膜中Galectin-3⁺小胶质细胞数量显著高于WT小鼠。miR-155cKO使APP/PS1小鼠的Galectin-3⁺细胞数减少了59%(图2g)。
- 然而,Western blot检测全视网膜Galectin-3蛋白水平在各组间无显著差异,提示Galectin-3的表达变化主要发生在特定的小胶质细胞亚群(MGnD)中,而非全视网膜水平。
- 稳态小胶质细胞标记物变化:
- P2ry12表达上调:Western blot显示,miR-155cKO使8月龄APP/PS1小鼠视网膜中P2ry12蛋白表达水平显著增加1.5倍(图2l, *p<0.05)。P2ry12是稳态小胶质细胞的标志性分子,其上调表明miR-155敲除促进了小胶质细胞向稳态表型的恢复。
- Apoe⁺细胞动态变化:ApoE是MGnD表型的关键分子。研究发现,在4月龄(疾病早期)APP/PS1小鼠中,Apoe⁺小胶质细胞数量较WT组已显著增加(*p<0.05),而miR-155cKO可下调其数量;在8月龄时,仅在WT组中观察到miR-155cKO导致Apoe⁺细胞显著减少(图2m)。这表明miR-155对Apoe的调控具有年龄和基因型依赖性。
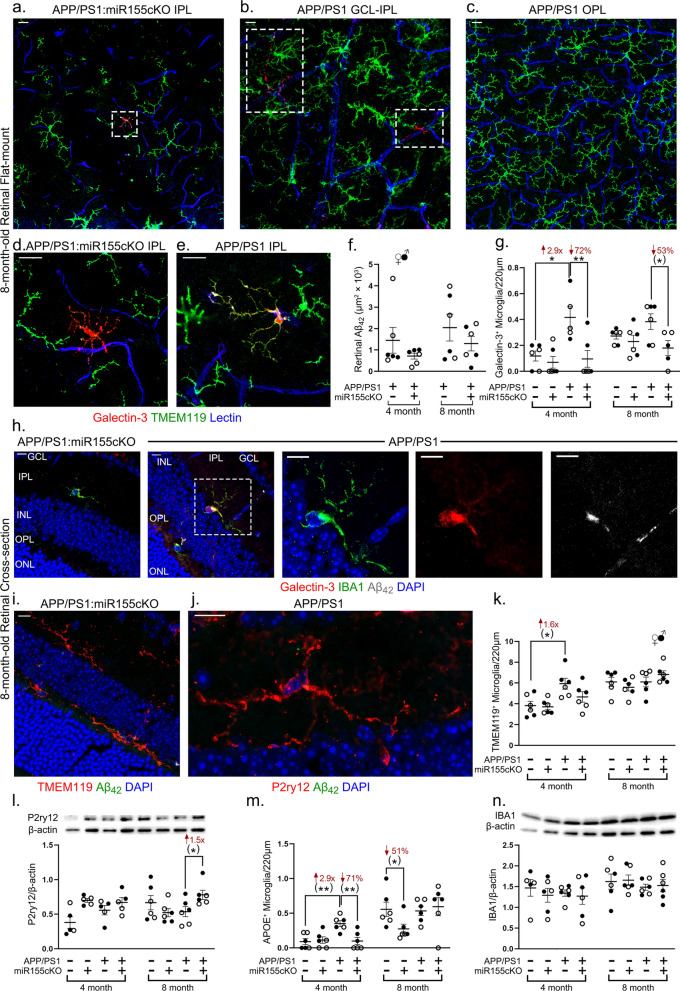
3. miR-155敲除抑制视网膜炎症反应
- 关键炎症介质下调:Western blot分析显示,miR-155cKO显著降低了APP/PS1小鼠视网膜中:
- NF-κB p65亚基的磷酸化水平(↓45%, 图3a, *p<0.05),表明抑制了促炎转录因子NF-κB的激活。
- 成熟IL-1β蛋白水平(↓40%, 图3b, *p<0.01)。
- TNF-α蛋白水平(↓38%, 图3c, *p<0.05)。
- 细胞因子谱改变:MSD多重检测进一步证实了miR-155敲除的抗炎效应:
- 在4月龄APP/PS1:miR-155cKO小鼠中:IL-6水平显著降低(↓52%, 图3h, *p<0.01)。
- 在8月龄APP/PS1:miR-155cKO小鼠中:抗炎细胞因子IL-10水平显著升高(↑2.1倍, 图3i, *p<0.05)。
- 这些结果综合表明,靶向小胶质细胞miR-155能有效抑制AD视网膜中关键的促炎信号通路(如NF-κB)和细胞因子(如IL-1β, TNF-α, IL-6)的产生,同时促进抗炎因子IL-10的表达,从而缓解视网膜神经炎症。
 图三
图三
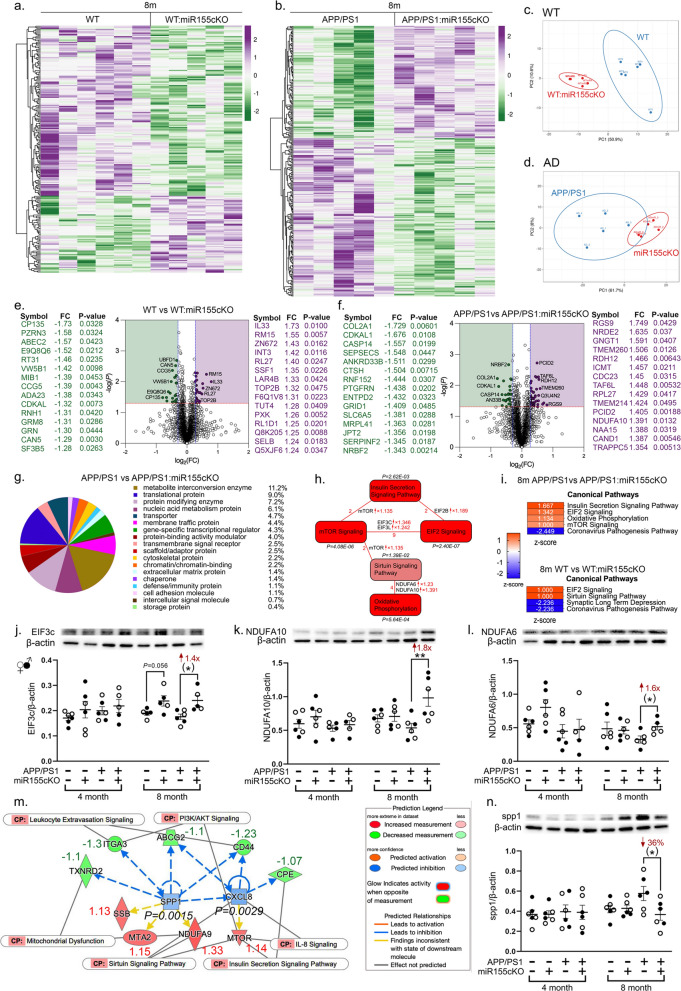
4. 蛋白质组学揭示PI3K-Akt信号通路激活 对8月龄小鼠视网膜进行TMT标记定量蛋白质组学分析,揭示了miR-155cKO调控的分子网络:
- 差异表达蛋白(DEPs):在APP/PS1:miR-155cKO vs APP/PS1比较组中,共鉴定出124个显著上调蛋白和87个显著下调蛋白(FC≥1.2, p<0.05)(图4b)。热图和主成分分析(PCA)清晰展示了不同组别间独特的蛋白表达谱(图4a, c, d)。火山图突出了变化最显著的DEPs(图4e, f)。
- 通路富集分析:IPA分析显示,miR-155cKO在APP/PS1小鼠视网膜中显著激活了多条保护性通路:
- PI3K-Akt信号通路相关通路:胰岛素分泌(Z=2.3)、mTOR信号(Z=2.1)、EIF2信号(Z=1.9)和sirtuin信号通路(Z=1.8)均被显著激活(图4h)。
- 氧化磷酸化(OXPHOS)信号通路也得到改善(Z=1.7, 图4i)。这些通路与细胞存活、代谢、蛋白质合成和线粒体功能密切相关。
- 关键分子验证:Western blot对蛋白质组学预测的关键上调分子进行了验证:
- EIF3c(mTOR和EIF2信号通路关键组分)表达上调1.8倍(图4j, *p<0.05)。
- NDUFA10(线粒体复合物I组分,sirtuin和OXPHOS通路相关)表达上调1.7倍(图4k, *p<0.01)。
- NDUFA6(线粒体复合物I组分)表达上调1.5倍(图4l, *p<0.05)。
- 上游调控因子预测:IPA预测miR-155cKO导致促炎因子Spp1(骨桥蛋白)和Cxcl8(IL-8)显著下调(图4m)。Western blot证实了视网膜中SPP1蛋白水平在cKO后显著降低(图4n)。免疫荧光显示SPP1主要表达于视网膜神经节细胞。
- 蛋白质组学结果从系统层面阐明,miR-155敲除通过激活PI3K-Akt-mTOR-EIF2-sirtuin这一核心保护性信号轴,并抑制Spp1等促炎因子,共同协调了其对视网膜的保护作用,特别是改善了线粒体功能。
 图四
图四
5. 靶向miR-155修复血-视网膜内屏障并减轻血管淀粉样病变
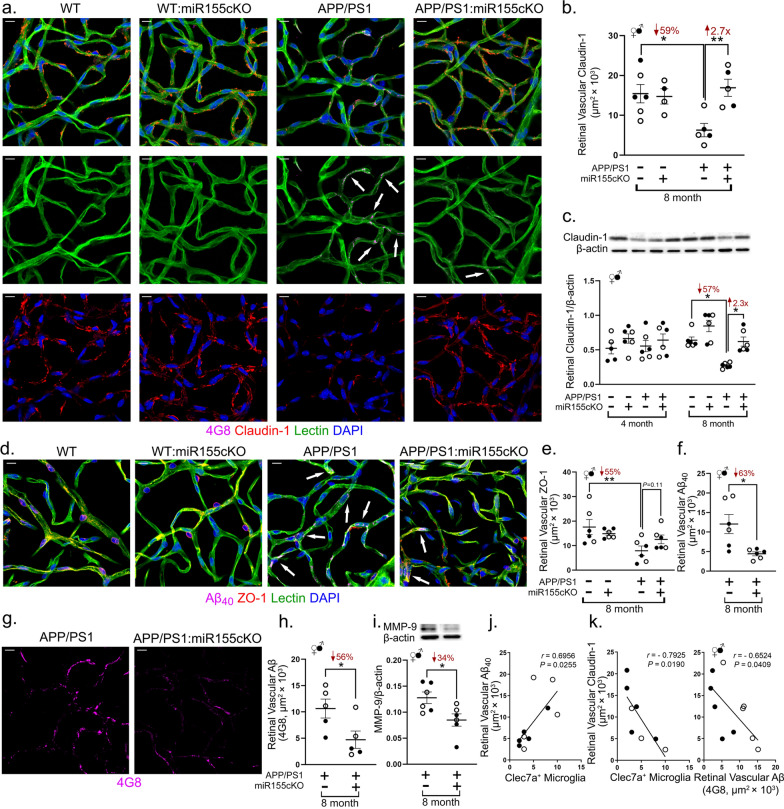
- 紧密连接蛋白恢复:
- 对分离的视网膜血管网络进行免疫荧光染色显示,APP/PS1小鼠血管 Claudin-1 免疫反应性显著降低,而miR-155cKO有效恢复了其表达(图5a)。立体学定量分析证实,cKO使视网膜血管Claudin-1免疫反应性增加了2.3倍(图5b, *p<0.01)。
- Western blot检测全视网膜Claudin-1蛋白水平,同样显示APP/PS1组降低,而cKO使其水平显著升高1.9倍(图5c, *p<0.05)。
- 虽然ZO-1在血管上的表达变化趋势类似(图5d, e),但未达到统计学显著性。这些结果表明miR-155cKO能有效修复AD视网膜中受损的iBRB,Claudin-1是关键靶点。
- 血管淀粉样病变减轻:
- 免疫荧光染色显示,APP/PS1小鼠视网膜血管存在明显的Aβ沉积(4G8⁺和11A50-B10⁺)。miR-155cKO显著降低了血管Aβ负荷:
- 4G8⁺总Aβ负荷↓44%(图5h, *p<0.01)。
- 11A50-B10⁺ Aβ₄₀负荷↓59%(图5f, *p<0.001)。
- 代表性图像清晰展示了cKO后血管Aβ沉积的减少(图5a, d, g)。
- 免疫荧光染色显示,APP/PS1小鼠视网膜血管存在明显的Aβ沉积(4G8⁺和11A50-B10⁺)。miR-155cKO显著降低了血管Aβ负荷:
- 酶学调控:Western blot检测显示,在APP/PS1小鼠视网膜中表达升高的基质金属蛋白酶9(MMP-9,一种可降解紧密连接蛋白的酶)在miR-155cKO后表达降低了37%(图5i, *p<0.05)。MMP-9的下调可能是iBRB修复的机制之一。
- 相关性分析:研究发现视网膜血管Aβ₄₀负荷与Clec7a⁺小胶质细胞数量呈显著正相关(r=0.78, p<0.001),而血管Claudin-1表达与Clec7a⁺细胞数量呈显著负相关(r=-0.69, p<0.01)(图5j-k)。这强烈提示Clec7a⁺ MGnD小胶质细胞与视网膜血管Aβ沉积和屏障损伤存在病理关联。
- 图解说明:小胶质细胞特异性敲除miR-155 (A) 产生双重效应:一方面减少促炎的Clec7a⁺/Galectin-3⁺ MGnD小胶质细胞亚群 (B),另一方面增加具有保护作用的P2ry12⁺稳态小胶质细胞 (C)。MGnD细胞的减少 (B) 直接导致促炎因子(如TNF-α, IL-1β)产生减少和NF-κB通路抑制 (D),进而减轻视网膜炎症 (F)。稳态小胶质细胞的增加或整体微环境改善 (C) 则激活了PI3K-Akt-mTOR-EIF2-sirtuin这一核心保护性信号轴 (E)。抑制炎症 (F) 和激活保护性通路 (E) 共同作用,上调了紧密连接蛋白(如Claudin-1)的表达 (G),从而修复了受损的血-视网膜内屏障 (H)。此外,MGnD细胞的减少 (B) 也与血管Aβ沉积的减轻直接相关 (I)。SPP1的下调也是该保护网络的一部分。
 图六
图六
讨论与意义
1. MGnD小胶质细胞在AD视网膜血管病变中的核心作用 本研究首次在AD模型小鼠视网膜中明确鉴定出Clec7a⁺和Galectin-3⁺ MGnD小胶质细胞亚群,并揭示了其病理意义:
- 空间定位与血管关联性:这些MGnD细胞主要定位于视网膜内层(GCL-IPL),并表现出与视网膜微血管的紧密黏附特性(图1d, 图2d)。这种空间定位强烈提示它们直接参与血管微环境的调节和病变过程。
- 与血管病理的定量关联:研究通过严谨的定量分析,建立了Clec7a⁺ MGnD细胞数量与视网膜血管病变的强相关性:
- Clec7a⁺细胞数量与血管Aβ₄₀负荷呈显著正相关(r=0.78, p<0.001)。
- Clec7a⁺细胞数量与血管紧密连接蛋白Claudin-1的表达呈显著负相关(r=-0.69, p<0.01)(图5j-k)。
- 这些数据为MGnD细胞直接参与AD视网膜血管淀粉样变性和屏障破坏提供了强有力的证据。
- Galectin-3的双重性与复杂性:虽然Galectin-3被广泛认为具有促炎和促进Aβ聚集的作用,但本研究观察到miR-155cKO后Galectin-3⁺细胞减少并未加重病理,且全视网膜Galectin-3蛋白水平未变。这提示Galectin-3在视网膜中的功能可能高度依赖于其细胞来源(MGnD小胶质细胞)和局部微环境,其具体角色需更深入研究。
2. miR-155作为治疗靶点的潜力与多效性调控机制 本研究发现小胶质细胞特异性敲除miR-155具有多方面的保护效应,其机制涉及多层次的调控:
- 多效性调控:miR-155缺失同时实现了抑制有害通路(TNF-α/NF-κB炎症轴)和增强保护性通路(PI3K-Akt-mTOR-EIF2-sirtuin)的双重目标。这种协同效应是其显著改善视网膜病变的基础。IPA预测及验证表明,其机制涉及:
- 上游调控因子Spp1(骨桥蛋白)的显著下调(IPA预测p=4.2E-6,WB验证,图4m, n)。SPP1是已知的促炎和趋化因子。
- 下游效应分子(如EIF3c, NDUFA10, NDUFA6)的协调上调,改善了蛋白质翻译和线粒体功能(图4j-l)。
- 转化医学价值:本研究为靶向miR-155治疗AD视网膜病变(及潜在的中枢病变)提供了坚实的临床前证据。锁定核酸(LNA)或类似的反义寡核苷酸(ASO)技术已被证明可有效实现miR-155的药理学抑制,这为未来临床转化提供了可行的技术路径。通过局部(如眼内给药)或系统性给药抑制miR-155,可能成为干预AD视网膜病变和神经炎症的新策略。
3. 视网膜作为AD早期诊断与监测窗口的价值 本研究结果进一步强化了视网膜作为AD病理“窗口”的重要性:
- 早期生物标志物:研究发现,在4月龄(症状前期)的APP/PS1小鼠视网膜中,就已出现小胶质细胞表型转换(如Apoe⁺细胞增加)和炎症因子谱的改变(如IL-6升高)。这提示视网膜小胶质细胞的变化可能早于明显的认知障碍或脑部严重病理,具有作为早期诊断生物标志物的潜力。
- 无创监测潜力:视网膜成像技术(如光学相干断层扫描血管成像OCTA、自适应光学成像)具有非侵入性、高分辨率和可重复性的优势。本研究发现的MGnD细胞(Clec7a⁺, Galectin-3⁺)通常位于浅层视网膜血管附近,理论上存在被特异性分子探针标记并通过先进成像技术进行在体无创监测的可能性。动态监测小胶质细胞表型转换将为评估AD进展和治疗反应提供新手段。
结论与展望
本研究首次在AD模型视网膜中鉴定出Clec7a⁺和Galectin-3⁺ MGnD小胶质细胞亚群,并系统性地证实了miR-155-Clec7a/Galectin-3轴是调控AD诱导视网膜血管病变的核心机制。通过他莫昔芬诱导的小胶质细胞特异性miR-155条件性敲除,成功实现了:
- 抑制MGnD表型:显著减少视网膜Clec7a⁺和Galectin-3⁺小胶质细胞数量。
- 促进稳态恢复:上调稳态标记物P2ry12的表达。
- 缓解神经炎症:抑制NF-κB活化,降低TNF-α、IL-1β、IL-6等促炎因子,升高IL-10。
- 激活保护通路:增强PI3K-Akt-mTOR-EIF2-sirtuin信号轴,下调SPP1。
- 修复血管屏障:上调紧密连接蛋白Claudin-1,恢复血-视网膜内屏障(iBRB)功能。
- 减轻血管淀粉样变:显著降低视网膜血管Aβ₄₀和总Aβ负荷。
这些效应共同作用,有效缓解了AD相关的视网膜血管病变和炎症。该研究不仅深化了对AD视网膜病理机制的理解,特别是小胶质细胞表型转换在血管损伤中的作用,也为开发基于调控小胶质细胞miR-155及其下游靶点(如Clec7a, Galectin-3, SPP1)的新型治疗策略(如miRNA抑制剂)提供了重要的实验依据。
未来研究方向包括:
- 人类样本验证:利用人死后AD患者视网膜样本,通过空间转录组或单细胞测序技术,深入解析人类AD视网膜中MGnD小胶质细胞亚群的存在、分子特征及其空间分布特征,并与小鼠模型结果进行对比。
- 转化研究:评估miR-155抑制剂(如LNA-antimiR-155)在更接近人类的灵长类AD模型或视网膜病变模型中的疗效和安全性,为临床试验奠定基础。探索局部眼内给药途径的可行性。
- 无创成像技术开发:研发能够特异性靶向并可视化视网膜MGnD小胶质细胞(如Clec7a, Galectin-3)的分子探针,结合OCTA、自适应光学等先进视网膜成像技术,实现AD病程中小胶质细胞表型动态转换的无创在体监测,评估其作为诊断和疗效监测生物标志物的临床价值。
- 机制深化:进一步研究miR-155下游具体靶分子(如RICTOR, SMAD1等)在介导MGnD表型转换和视网膜血管病变中的精确作用机制,以及不同MGnD亚群(Clec7a⁺ vs Galectin-3⁺)的功能差异。
- 治疗窗口探索:研究在AD不同阶段(早期 vs 晚期)进行miR-155干预的效果差异,确定最佳治疗时间窗。
总之,靶向小胶质细胞miR-155及其调控的MGnD表型,为治疗AD相关的视网膜病变乃至中枢神经系统病变开辟了充满希望的新途径。
| 名称 | 货号 | 规格 |
| hKLK3 DuoSet (1 KIT) | DY1344 | 1KIT |
| U-PLEX Metabolic 3-Plex Combo 2 (ms) SECTOR (5 PL) | K15305K-2 | 5PL |
| RNeasy Micro Kit (50) | 74004 | 50Test |

