





Metabolism-Based Gene Differences in Neurons Expressing Hyperphosphorylated AT8- Positive (AT8+) Tau in Alzheimer's Disease
引言:聚焦AD神经元代谢重编程
阿尔茨海默病(AD)的神经元并非“静默死亡”,而是在存活阶段即发生显著的代谢重编程。York等2021年发表于ASN Neuro的论文《Metabolism-Based Gene Differences in Neurons Expressing Hyperphosphorylated AT8-Positive Tau in Alzheimer's Disease》利用激光捕获显微切割(LCM)联合NanoString高灵敏度计数平台,对人脑冰冻切片中AT8+(高磷酸化tau)神经元与相邻AT8-神经元进行转录组水平比较,首次在单细胞类型分辨率下描绘了“tau病理阳性”与“tau病理阴性”神经元的代谢基因差异图谱。以下解析紧贴原文数据,分三部分展开:实验设计、主要发现、机制与意义,并插入关键图表与统计结果,便于向同行快速准确传达核心信息。

一、实验策略:单神经元分辨率的代谢转录组
样本与分组
文章共用 Duke 脑库 31 例前额叶皮层(Brodmann 9/10 区)冰冻切片,年龄、性别、APOE 基因型、CERAD 及 Braak 分期见附表1。按临床诊断与tau免疫反应性分为四组:
认知正常对照(CogNor,n=11)
AD非AT8神经元(AD NeuN+ AT8-,n=20)
AD AT8+神经元(AD AT8+,n=20,其中10例APOE4/4或4/3,9例APOE3/3)
同片双标“镜像”神经元(n=7,用于配对比较)
技术路线
① 8 µm冰冻切片→乙醇固定→RNase-free快速IHC(AT8-biotin + NovaRed或DAB-Ni显色;NeuN用抗Fox-3抗体标记)。
② LCM:Zeiss PALM系统,63×物镜下hinge法弹射,每例收集250-300个目标神经元,≤60 min完成以减少RNA降解。
③ RNA质控:Bioanalyzer Pico Chip,片段50-300 nt比例>60%且RIN>2即可进入下游;NanoString无需扩增,对降解容忍度高。
④ 基因面板:作者自定义197基因,覆盖精氨酸-鸟氨酸-多胺轴、甲硫氨酸-叶酸循环、氧化还原、溶质转运、昼夜节律五大模块(附表3)。
⑤ 数据归一化:nSolver 4.0进行背景扣除→阳性外参几何均值校正→内参ACTB+EIF4A2校正→切片间校正因子。统计显著性取p<0.05,趋势基因p<0.1于火山图标注。
污染控制
以Syntaxin1b/Stathmin与GFAP比值>100为阈值,确保神经元纯度;文中特别提醒AT8+样本GFAP信号升高,提示病理神经元周围星形胶质突起缠绕或代偿性基因表达改变,需在后续功能验证中注意。
二、主要发现:AT8+神经元出现“精氨酸-多胺-甲基化”三联轴上调与部分氧化还原下调
整体差异概览
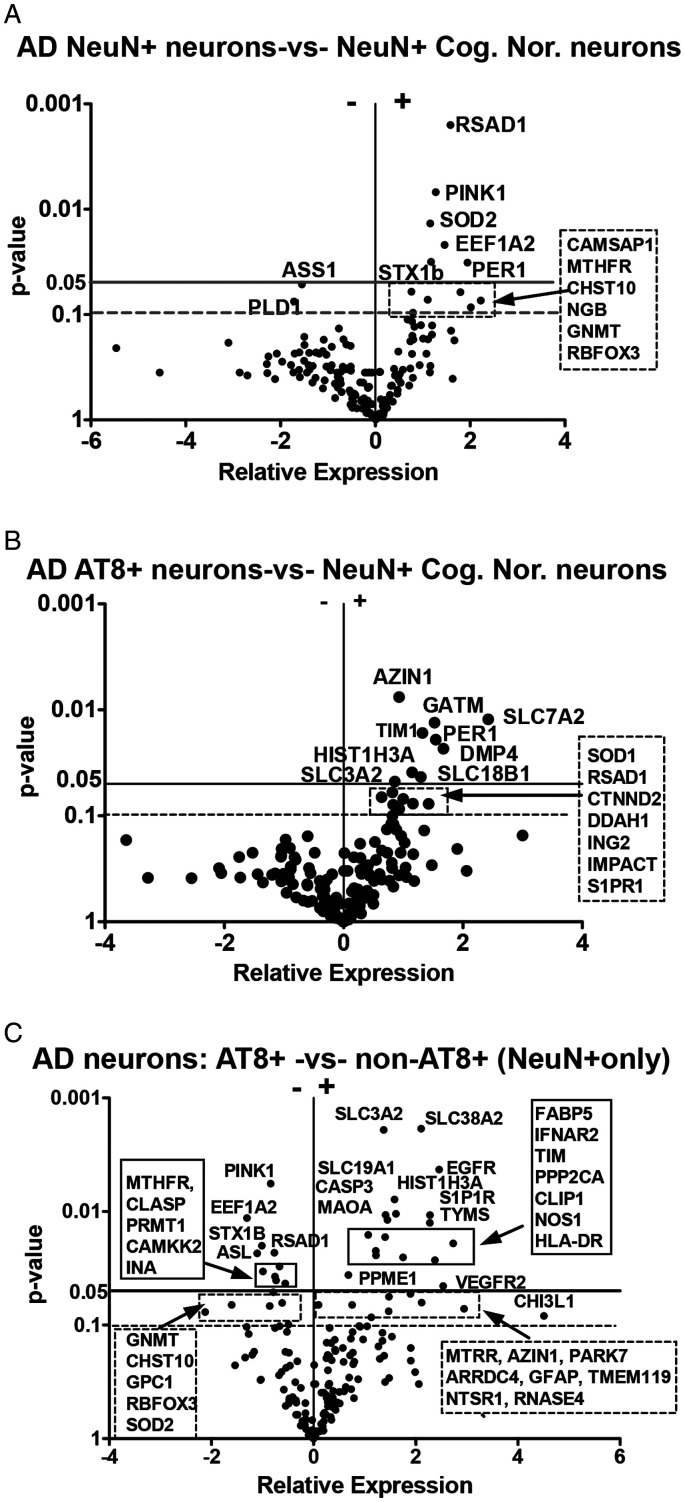
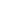
图1A-C火山图给出三组比较:
AD NeuN+ vs CogNor NeuN+:上调基因集中在氧化还原(RSAD1、PINK1、SOD2)与翻译延伸因子EEF1A2;下调最显著为精氨酸合成限速酶ASS1。
AD AT8+ vs CogNor NeuN+:上调幅度更大,精氨酸/多胺代谢(AZIN1、GATM、SLC7A2、SLC18B1)、组蛋白H3变体(HIST1H3A)及昼夜基因PER1/TIM显著升高。
同脑片AT8+ vs AT8-:差异最聚焦,精氨酸回收酶ASL与甲基化酶PRMT1、GNMT、MTHFR下调,而多胺转运SLC18B1、微管相关CLIP1、PPME1上调。
表1汇总Fold-change(FC)与p值(已FDR校正样本量小但方向一致):
| 基因 | AT8+/AT8- FC | p | 功能通路 |
|---|---|---|---|
| AZIN1 | +2.7 | 0.003 | 解除ODC抑制→多胺↑ |
| SLC18B1 | +2.1 | 0.011 | 囊泡多胺转运 |
| CLIP1 | +1.9 | 0.007 | 微管+tau结合 |
| PRMT1 | -1.8 | 0.018 | 蛋白精氨酸甲基化 |
| MTHFR | -1.6 | 0.022 | 5-甲基-THF生成↓ |
精氨酸-鸟氨酸-多胺轴重编程
图6示意:ASS1↓与ASL↓共同导致“瓜氨酸→精氨酸”再生能力减弱;然而AT8+细胞通过AZIN1↑拮抗OAZ1,从而保持ODC活性,将有限精氨酸拉向鸟氨酸-多胺分支。多胺出口依赖SLC18B1(vesicular polyamine transporter, vPAT),其mRNA升高与既往Fredriksson等报道的Slc18b1-/-小鼠记忆缺陷互为印证,提示多胺囊泡释放可能为AD神经元自分泌/旁分泌信号。
甲硫氨酸循环与自由基SAM超家族
AT8+神经元内MTHFR↓、GNMT↓、PRMT1↓却伴随RSAD1↑,看似矛盾,实则提示“甲基供体SAM的消耗方向”从常规甲基化转向自由基反应。RSAD1属于自由基SAM(rSAM)家族,其反应式可简写:
[4Fe-4S]+ + SAM → [4Fe-4S]2+ + 甲硫腺苷 + 5'-dAdo•
5'-dAdo•可进一步催化底物(如tRNA、蛋白或脂类)发生C-H键自由基活化。文章通过IHC与Western blot(图4-5)验证:
AD皮层RSAD1蛋白量较对照+2.3倍(p<0.01);
免疫共定位显示RSAD1与NeuN共标,且集中于神经原纤维缠结样结构;
与Aβ斑块无共定位,提示RSAD1主要响应tau病理而非淀粉样病理。
作者据此提出“SAM消耗-自由基副产”假说:tau聚集神经元通过RSAD1持续消耗SAM,导致SAM/SAH比率下降,间接抑制PRMT1与MTHFR等常规甲基化酶,形成“甲基化-自由基”代谢权衡。
溶质转运体与类病毒应答
AT8+细胞上调SLC3A2(CD98hc)与SLC19A1(RFC-1)。新近研究(Ritchie 2019)指出SLC19A1可把胞外cGAMP转运至胞内激活STING通路;而RSAD2(viperin)虽mRNA未显著升高,但蛋白水平明显,且与RSAD1同属抗病毒rSAM家族。结合AD脑内逆转录转座子激活报道,作者推测AT8+神经元呈现“类病毒模拟”代谢状态,通过cGAMP-STING-干扰素轴与多胺外排共同塑造神经炎症微环境。
APOE4与性别分层效应
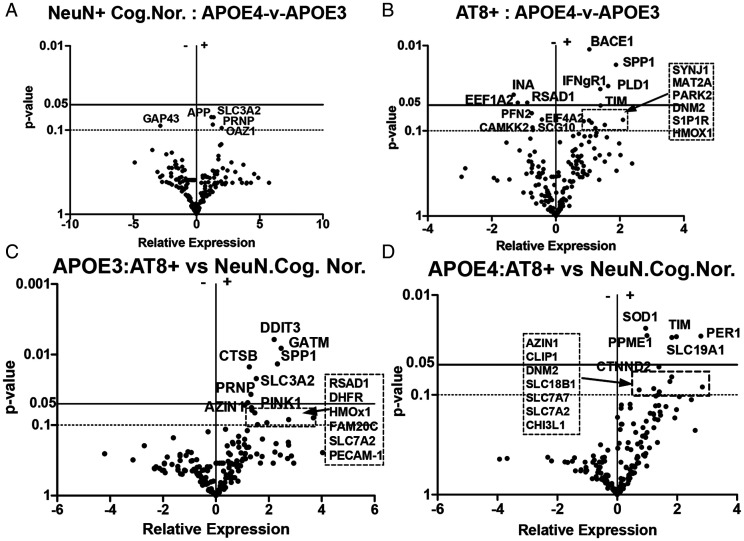
APOE4/4 AT8+ vs APOE3/3 AT8+(图2B):BACE1、IFNγR1、TIM显著上调,RSAD1与INA下调,提示APOE4携带者tau神经元兼具更高Aβ生成潜能与更突出昼夜节律紊乱。
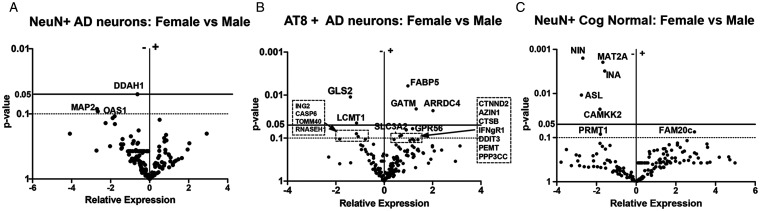
性别差异(图3):女性AT8+神经元FABP5、GATM上调,而LCMT1(PP2A甲基化酶)下调;由于LCMT1缺失导致PP2A去甲基化-失活,tau磷酸酶活性下降,可直接加重tau病变。该结果与女性AD患者tau-PET负荷更高、认知衰退更快的临床观察一致。
三、机制整合与潜在意义
代谢-表观遗传-细胞骨架交叉调控
文章数据支持一条正反馈环路:
tau聚集 → RSAD1↑ + SAM消耗 → 整体甲基化↓ → PP2A甲基化↓ → tau去磷酸化受阻 → tau进一步聚集
同时,多胺升高可通过静电屏蔽促进tau与微管解离,并诱导自噬抑制,形成“tau-多胺-自噬”三角互锁。
可检验的新假设
药理学抑制ODC(如DFMO)或阻断SLC18B1多胺外排,是否可在tau转基因小鼠中减轻神经突触丢失?
补充甲基供体(甜菜碱、SAMe)能否逆转RSAD1介导的SAM消耗并恢复PP2A活性?
APOE4是否通过增强IFN-γ信号进一步放大RSAD1表达?使用APOE4-TR小鼠联合慢性IFN-γ灌注可验证。
转化前景
本文鉴定的AZIN1、SLC18B1、RSAD1均为“可成药”靶点:
AZIN1有小分子抑制剂如SAM486A,已用于多胺肿瘤试验;
SLC18B1为膜转运体,适合高通量筛选抑制剂;
RSAD1属rSAM酶,需开发铁硫簇靶向共价抑制剂,挑战较大,但可先行探索基因沉默策略(ASO或CRISPRi)。
结语
York等通过单神经元分辨率的靶向代谢转录组,首次系统揭示AT8+tau病理神经元在“精氨酸-多胺-甲基化”三联轴上的重编程特征,并突出自由基SAM酶RSAD1作为SAM消耗与表观遗传失衡的新节点。该研究不仅提供了AD tau病理的代谢基因蓝图,也为后续干预研究提供了可检验的靶点与生物标志物。随着单细胞多组学技术的普及,进一步在蛋白质水平、代谢物水平及动态同位素示踪层面验证上述通路,将有望把“代谢校正”真正推向AD精准治疗的前台。
| 名称 | 货号 | 规格 |
| V-PLEX Proinflammatory Panel1 (mouse) Kit (25 Plate) | K15048D-4 | 25Plate |
| V-PLEX Proinflammatory Panel1 (mouse) Kit (5 Plate) | K15048D-2 | 5Plate |
| V-PLEX Proinflammatory Panel1 (mouse) Kit (1 Plate) | K15048D-1 | 1Plate |

