此篇文章摘录于Methods in Molecular Biology 2032中的一篇,名为Fluorescent Cell Barcoding for Immunophenotyping(10.1007/978-1-4939-9650-6_3)(IF: 38.31)其作者是Trabanelli Sara , Gomez-Cadena Alejandra , Jandus Camilla。
荧光细胞条形码(FCB)是一种在抗体染色前使用不同浓度的不同荧光染料对细胞进行标记从而实现高通量多重检测方法,是一种对流式细胞术(FCM)的复用技术。单个样品被唯一标记,然后混合在一起,进行染色并作为单个样品进行分析, 这样可以减少技术误差并提高检测通量和获取速度。
一、 简介及原理
流式细胞术分析过程的标准化是非常重要的,这样可以获得可重复的高质量数据,提高在实验室内和实验室间水平随时间的可比性。荧光细胞条形码(FCB)通过将多个样品混合在一个试管中进行进一步染色和分析,从而实现高通量多参数流式细胞术。实验使用不同浓度的共价结合荧光染料进行细胞标记,因此样品被打上“条形码”,并根据荧光发射波长和强度进行区分每种细胞。多种打上“条形码”的样本可以混合在一起,然后在同一个试管中用抗体进行染色、上机、分析等流程。因此,FCB最大限度地减少染色变异性,抗体消耗和所需的总样本量。免疫细胞FCB实验可以在新鲜全血、外周血单个核细胞(PBMC)或冷冻保存的PBMC上进行表型分析。然而,FCB的实现需要更加优化,本文介绍对FCB实验流程及注意事项。
二、试剂

△点击放大图片
三、实验方法
1、样品准备
淋巴细胞表型的FCB可以使用新鲜的样本如全血或外周血单个核细胞(PBMCs)或冷冻保存的PBMCs。如果使用冻存的PBMCs,在37 ℃的水浴中解冻细胞1-2分钟。加入2 mL PBS ,400g离心5分钟。丢弃上清液,旋涡使细胞团分散。如使用的是新鲜的PBMCs,请从步骤5开始。
本实验使用新鲜全血血液:
(1) 在抗凝血剂(EDTA或肝素钠)存在的情况下收集至少0.2 mL全血。
(2)用1:10 Ack Lysing buffer裂解。取0.2 mL新鲜血液,加入2 mL Ack Lysing buffer,在冰上孵育10分钟(见注意事项3)。
(3)400g离心5分钟,弃去上清液。
(4)重复步骤2和3。
(5)加入3mL 1x BD Phosflow Lyse/Fix Buffer,用移液法轻轻混合,室温孵育15分钟。
(6)400g离心5分钟。丢弃上清液,旋涡使细胞球团分散开。
(7)加入3mL预冷1x BD Perm Buffer II。在冰上孵育20分钟(见注意事项4)。
(8)加入2 mL PBS清洗,以400g离心5分钟。丢弃上清液,旋涡使细胞球团变松。
(9)将细胞在低温1x BD Perm Buffer II中重悬,并将细胞置于冰中直到使用。
每管的体系为30 μL,细胞量为 0.6-1x106 。另外,需要设置一管未染色样品管;每种染料单染管(如DyLight 800,DyLight 350或者Pacific Orange)。
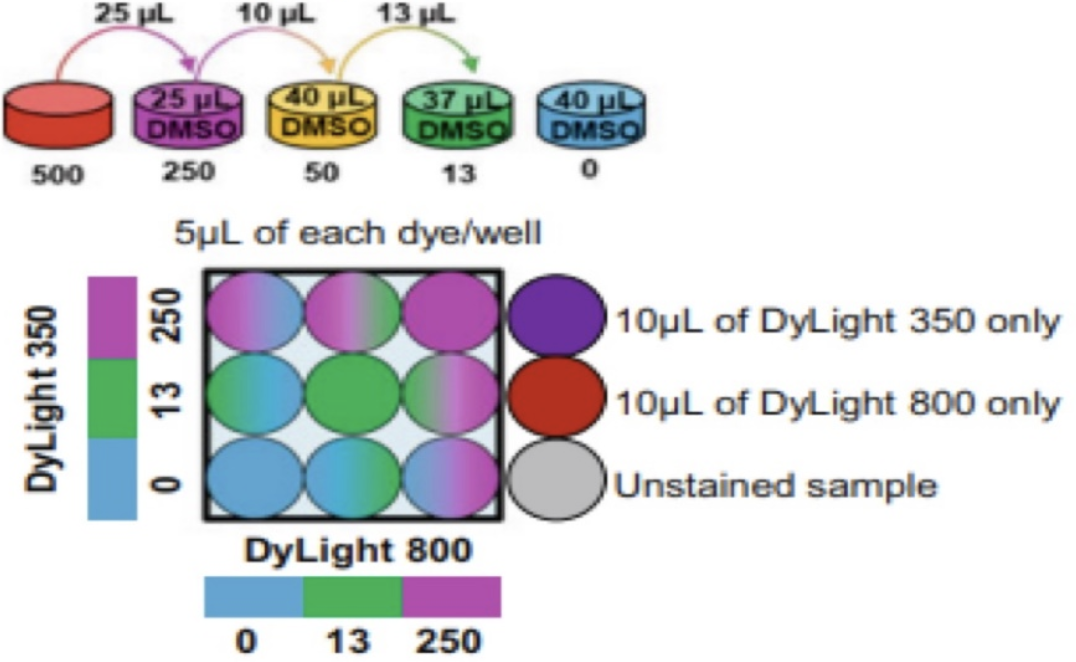
2、9样品矩阵实验操作(3x3)
在实验开始前,使用DMSO将染液保存液稀释到工作液,因为DMSO容易沉淀。
(1)避光室温下解冻500 μg/mL染料原液。解冻后,大力涡旋。
(2)在圆底96孔板中用DMSO配制染料工作浓度,并将孔标记为“DyL350-0”、“DyL350-13”、“DyL350-250”和“DyL800-0”、“DyL800-13”、“DyL800-250”(见注意事项5)。
每种染料配制如下:
250μg/mL稀释液=25 μL 500 μg/mL稀释液+ 25 μL DMSO
50μg/mL稀释液=10 μL 250稀释液+ 40 μL DMSO
13μg/mL稀释液=13 μL 50稀释液+ 37 μL DMSO
0μg/mL稀释液=30 μL DMSO
50稀释液仅用于13稀释液的制备,不用于FCB矩阵制备(见注意事项6)。
(3)如图1所示,将稀释后的染料5 μL加入96孔圆底板的适当孔中。当最终体积为40 μL/孔(10 μL染料+30 μL样品)时,最终浓度为0、3.25和62.5 μg/mL。
(4)密封或盖上盘子,并在室温下避光保存。
(5)转到进行荧光细胞条形码步骤。
△点击放大图片
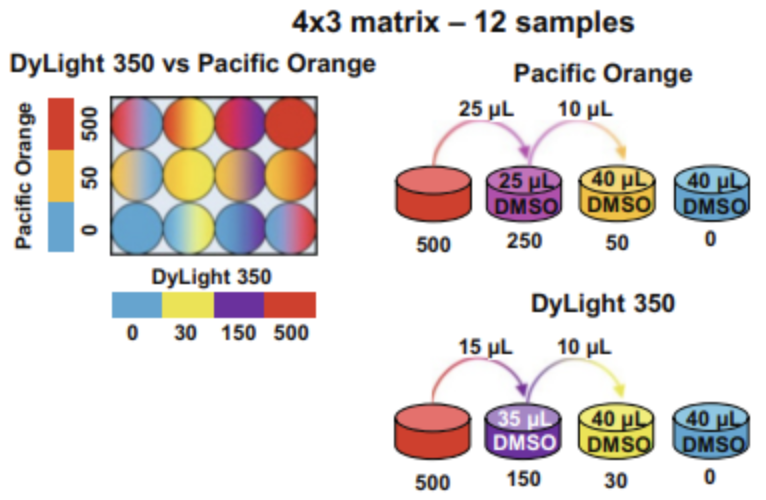
3、12种样品矩阵实验操作(4x3)
在实验开始前,使用DMSO将染液保存液稀释到工作液,因为DMSO容易沉淀
(1)避光室温下解冻500 μg/mL染料原液。解冻后,大力涡旋。在一个圆底96孔板中,用DMSO按如下方法制备染料工作浓度(图2)。
(2)对于DyLight 350,管的名称如下:“DyL350-0”,“DyL350-30”,“DyL350-150”和“DyL350-500”。准备稀释如下:
500μg/mL稀释液=50 μL 500 μg/mL稀释液
150μg/mL稀释液=15 μL 500 μg/mL稀释液+35 μL DMSO
30μg/mL稀释液=10 μL 150稀释液+40 μL DMSO
0μg/mL稀释液=40 μL DMSO
当染料和样品的体积为40 μL/孔时,DyLight 350的最终浓度分别为0、3.8、18.8和125μg/mL
(3)对于Pacific Orange,将管标为“PO-0”、“PO-50”和“PO-500”。
准备稀释如下:
500μg/mL稀释液=50 μL 500 μg/mL的稀释液
250μg/mL稀释液=10 μL 500 μg/mL稀释液+ 10 μL DMSO
50μg/mL稀释液=10 μL 150稀释液+ 40 μL DMSO
0μg/mL稀释液= 40 μL DMSO
250μg/mL稀释液只用于准备50μg/mL稀释液,不用于实验。体积为40 μL/孔。染料和样品在12个样品矩阵中结合后,PO最终浓度为0、6.3和125 μg/mL。
(4)将稀释后的染料5 μL加入96孔圆底板的适当孔中。
(5)密封或盖上盘子,并在室温下避光保存。
(6)转到进行荧光细胞条形码步骤
△点击放大图片
4、荧光细胞条形码
荧光矩阵和样品准备好,按如下步骤进行FCB。除非实验有特别要求,否则在RT中进行所有步骤。
(1)每种样品按矩阵位置分别加入30 μL。充分混合溶液,因为染料容易沉淀。
(2)将30 μL样品加入10 μL DyLight 350的一个孔中,另将30 μL样品加入10 μL DyLight 800的另一个孔中。这些样品在样本准备步骤(9)中操作,将用作流式设置的单色对照。
(3)密封,在冰上孵育15-25分钟。全血孵育时间较长为宜(见注意事项7)。
(4)将所有9个条形码样本转移到一个名为“Combo”的FACS管中。将步骤(2)中制备的单色对照转移到两个单独的标有“DyL350”和“DyL800”的FACS管中。
(5)用3ml 1x Barcoding Wash Buffer (见注意事项8)。400g离心5分钟,丢弃上清液。旋涡使细胞团分散。重复步骤(5)(参见注意事项9)。
(6)将“Combo”、“DyL350”和“DyL800”重悬在300 μL的1x Barcoding Wash Buffer中。将“DyL350”和“DyL800”冷藏至上机。
(7)按照说明书将抗体添加到相应的试管中,在黑暗中RT孵育20-30分钟。
(8)用3mL 1x Barcoding Wash Buffer清洗。400g离心5分钟,然后丢弃上清,旋涡使细胞球团分散,再重复一次洗涤步骤。
(9)将细胞重悬于300-500 μL 1x Barcoding Wash Buffer中,冷藏在4℃,直至上机。
(10)可以在细胞仪上使用相应的激光(355、407或630 nm)进行采集。荧光补偿可以使用荧光补偿微球和已经使用最高浓度染料进行条形码标记的细胞进行计算。至少需要记录5万个淋巴细胞。
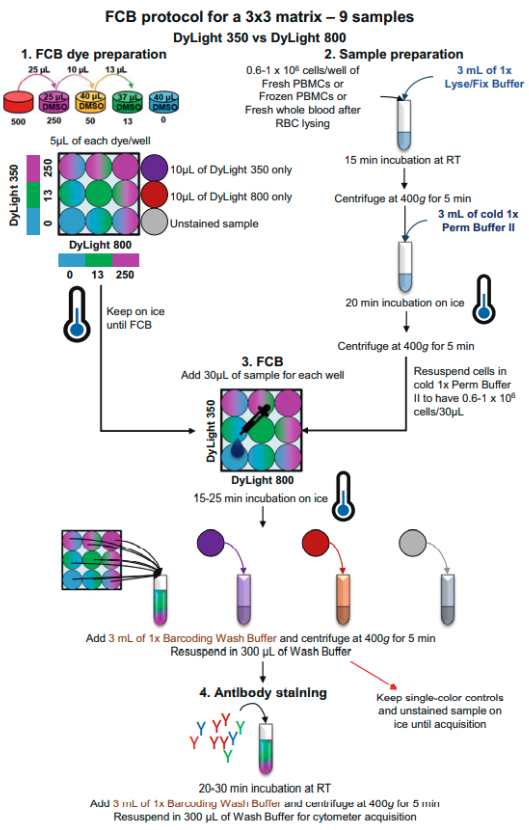
△点击放大图片
5、对照样本设置
在方案优化过程中,使用一个矩阵中的桥式样品来测量分析内的变异性。事实上,阳性细胞的百分比和MFI值在同一矩阵中的所有9/12条形码群体中不应该有显著差异
对照样本包括不固定破膜和FCB对照和破膜固定但不FCB对照。这两个对照用于比较条形码前和条形码后的阳性细胞百分比。
5.1 不固定破膜以及FCB对照
(1)将样本制备步骤4中的细胞在PBS中重悬。
(2)根据说明加入抗体,在黑暗中RT孵育20-30分钟。
(3)用3ml PBS清洗,400 g离心5分钟,弃上清。旋涡使细胞颗粒松动。
(4)如果需要进行细胞内染色,加入200 μL CytoFix/ CytoPerm BD buffer, 4℃孵育30分钟。其他方面跳到步骤7。
(5) 用2mL 1x Perm Wash,400克离心5分钟,丢弃上清液,旋涡使细胞球团松散。
(6)将细胞重悬于175 μL (1x Perm Wash)中,并胞内抗体。在4℃的环境中在黑暗中孵育30-40分钟。
(7)用2 mL 1x Perm Wash。400g离心5分钟,弃上清,旋涡使细胞球团散开。
(8)将细胞重悬于300 μL 1x Perm Wash中
5.2 破膜固定不FCB对照
(1)按照样本处理步骤1-4准备细胞。
(2)离心后,加入3 mL的1x Lyse/Fix Buffer, RT下孵育15分钟。
(3)400 g离心5分钟。丢弃上清液,旋涡使细胞球团变松。
(4)加入3 mL冷1x Perm Buffer II。在冰上孵育20分钟。
(5)加入2 mL PBS,以400 g离心5分钟。丢弃上清液,旋涡使细胞球团变松。
(6)将细胞重悬于200 μL的1x Barcoding Wash Buffer液中。
(7)将抗体添加到适当的试管中,在黑暗中RT孵育20-30分钟。
(8)用3 mL 1x Barcoding Wash Buffer。400 g离心5分钟,然后丢弃上清,旋涡使细胞球团散开。再重复一次洗涤步骤。
(9)将细胞重悬于300-500 μL 1x Barcoding Wash Buffer中,冷藏在4 ℃,直至上机。
为了测量跨矩阵和时间的分析间变异性,我们建议在每个矩阵中包括一个用于优化FCB流程的样本。该样本将被用作内部控制和桥梁,可以促进不同的实验间的纵向研究。这种桥状样品应该用相同的染料浓度对细胞打条形码,从而在矩阵中保持相同的位置。通过比较跨矩阵的桥式样本的阳性细胞和/或MFI值的百分比,可以计算分析间的变异性,通过异常值可以找出需要重复的实验。
6、采集后分析
采集后数据分析可使用FlowJo或RStudio软件进行。下面描述了使用这两种软件的推荐分析步骤:
6.1FlowJo
在FlowJo软件中,可以对条形码样品进行流式细胞术分析或反卷积分析,如下所示:
(1)制作采集后补偿矩阵,并应用于条形码样本,导出矩阵为.csv文件。
(2)根据前向散射区(FSC-A)和侧向散射区(SSC-A)识别淋巴细胞和/或单核细胞。
(3)基于FSC-A vs FSC-H排除黏连细胞。
(4)在单个细胞上,识别基于FCB染料的条形码群体(例如,DyLight 800 vs DyLight 350)。
(5)在每个条形码群体上,根据抗体染色执行门控策略。

△点击放大图片
6.2 RStudio
RStudio也可以使用以下包(flowCore,flowClust, flowViz, flowWorkspace, ggcyto和flowType)。在RStudio中,细胞仪荧光通道可以识别抗体 (例如,在LSR Fortessa细胞仪上,使用BUV396-A通道获取DyLight 350,使用APC-H7-A通道获取DyLight 800)识别)。
(1)通过使用rectangleGate功能和FSC-A和SSC-A参数过滤数据来清除碎片。使用子集方法删除FSC值小于20 k的事件。
(2)利用FlowJo分析第1步制作的矩阵和flow Workspace的补偿功能对数据进行补偿。
(3)使用arcsinh函数转换补偿数据。
(4)FCB染料通道的过滤数据(例如BUV396-A和APC-Cy7-A)由矩形函数。
(5)使用flowClust函数识别条形码种群。集群启动是通过在FCB染液通道上设置varNames参数来执行的(例如,BUV396-A和APC-Cy7-A), K根据矩阵中条形码样本的数量(9或12)设置。
(6)聚类完成后,可以使用ggcyto包显示数据。
(7)条形码群体被单独识别,并为进一步分析创建一个流集。
(8)门控策略可以使用矩形函数执行,使用感兴趣的参数(例如,CD3 vs SSC-A或CD4 vs CD8),并使用ggcyto同时显示包含在一个流集中的条形码群体。
7、推荐评估FCB的有效性和检测内/检测间的可变性
7.1 MFI倍增计算
反卷积纯度表示为MFIs和CV之间的距离,以获得70%各自编码的群体,纯度为95%。另一些文章称,为了获得良好的反卷积,MFIs应该以三倍的增长来分离。
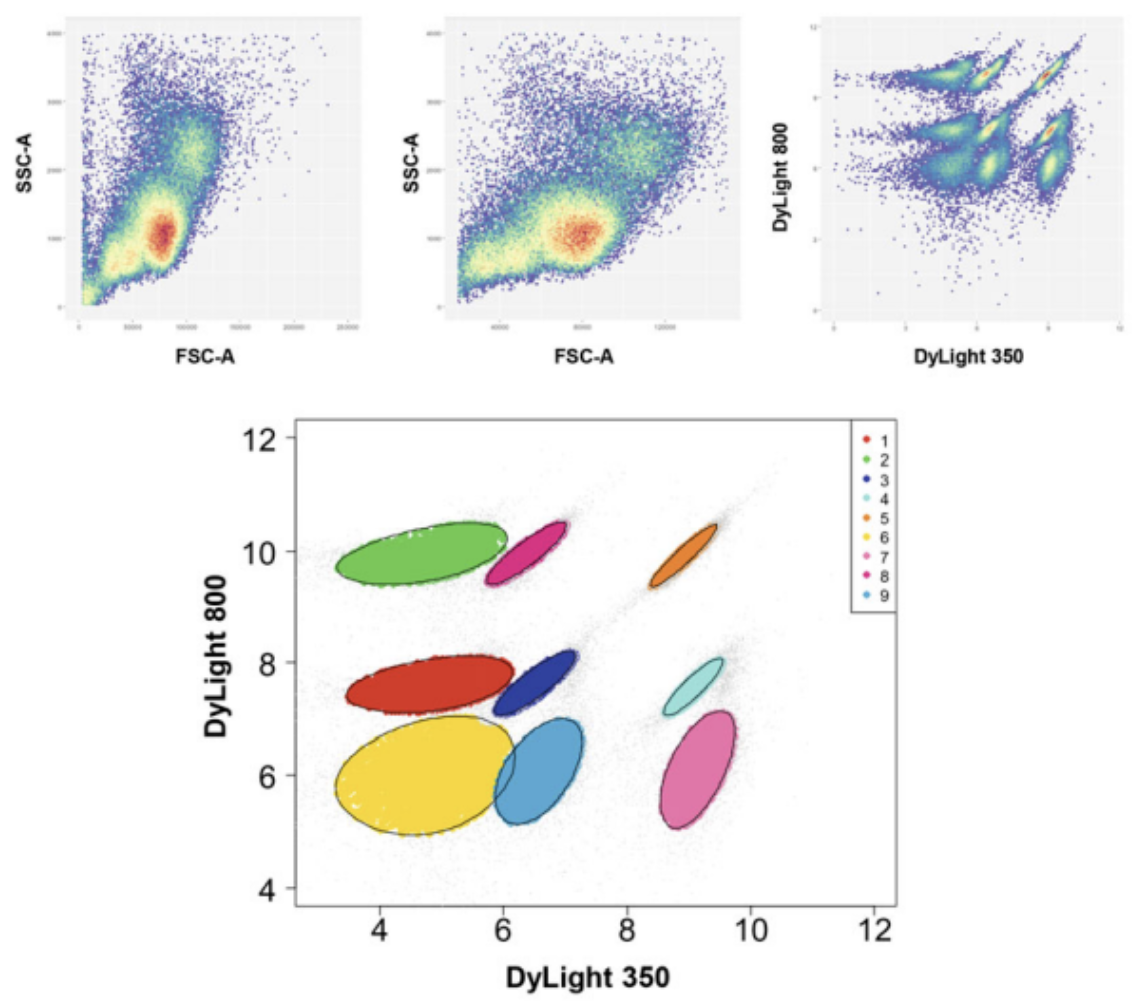
△点击放大图片
(1)在FlowJo软件中制作采集后补偿矩阵,并应用于条形码样品。
(2)根据前向散射区(FSC-A)和侧向散射区(SSC-A)识别淋巴细胞和/或单核细胞。
(3)排除基于FSC-A vs FSC-H的双态。
(4)在单个细胞上,以直方图的形式显示每种FCB染料,并识别每种使用的染料浓度,并相应地命名群体(例如,当制作9种样品矩阵时,“DyL350-0”,“DyL350-13”,“DyL350-250”和“DyL800-0”,“DyL800-13”,“DyL800-250”)。
(5)计算每种FCB染料稀释的MFI和CV,导出数据为.csv或.xls文件。
(6)在电子表格中,计算MFI折叠变化如下:
MFI fold increase =[MFIpeak2-CVpeak2]/[MFIpeak1 +CVpeak1]
7.2检测内/检测间阳性细胞百分比的变异性计算
(1)使用FlowJo软件计算对照样品中阳性细胞(如CD3+细胞,CD4+细胞)的百分比,并将数据导出为.csv或.xls文件。
(2)计算可变性的范围为<对照组的平均值-2SD或>对照组的平均值+2SD。
(3)比较反卷积后条形码总体与匹配对照样本的百分比。值应该在±2SD之内。
(4)对于每个细胞群(如CD3+细胞),计算一个基质中所有条形码样本的平均值,并与该基质中使用的所有匹配对照样本的平均值进行比较。数值应在合理范围内控制±2SD。
(5)对于每个细胞群(如CD3+细胞),计算所有基质中相同FCB染料稀释度(如0 μg/mL)的所有条形码样品的平均值,并与使用的所有匹配对照样品的平均值进行比较。数值应在±2SD的控制范围内。
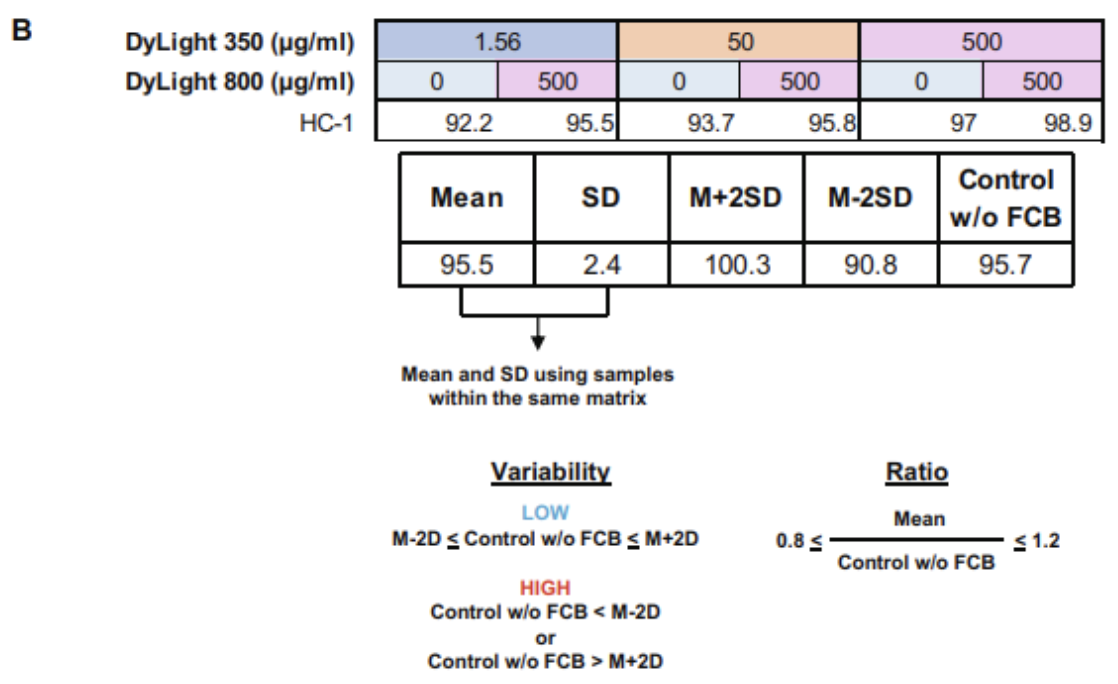
△点击放大图片
7.3 计算每种荧光素测定内和测定间MFI的变异性
1. 使用FlowJo软件计算条形码样本中使用的每个抗体的MFI,并将数据导出为.csv或.xls文件。
2. 计算可变性的范围为<对照组的平均值-2SD或>对照组的平均值+2SD。
3.将反卷积后的条形码种群的MFI值与最低浓度的条形码种群(例如,0 μg/mL)进行比较(见注释10和11)。值应该在2SD之内。
4. 对于每个细胞群(如CD3+细胞),计算一个基质中所有条形码样本的平均值,并与该基质中使用的所有匹配对照样本的平均值进行比较。数值应在合理范围内
控制 2 SD。
5. 对于每个细胞群(如CD3+细胞),计算所有基质中相同FCB染料稀释度(如0 μg/mL)的所有条形码样品的平均值,并与使用的所有匹配对照样品的平均值进行比较。数值应在2SD的控制范围内。
四、注意事项
1. 我们使用DyLight 350和DyLight 800进行了FCB染料的组合,但用户可以根据细胞仪设置和使用的荧光染料选择不同的染料(例如,Pacific Orange)。实际上,如果选DyLight 350和DyLight 800,细胞仪需要配备紫外 (355 nm)和红色(633 nm)激光。如果没有紫外激光,我们建议使用Pacific Orange和DyLight 800对细胞打上条形码,因为它们是由紫色(407 nm)和红色(633 nm)激光激发的。然而,由于其他紫色通道的溢出,Pacific Orange限制了其他由紫色激光激发的荧光染料的使用。
2. 为避免重复解冻循环,可配制500 μg/mL原液。但是,不要做小体积的溶液,因为溶液不可能像大体积的溶液一样均匀和稳定。
3.使用移液器将样品与裂红液轻轻混合,在孵育期间偶尔重复。
4. 如果pSTATs包含在抗体染色中,对于渗透性比对,使用3 mL冷Perm Buffer III而不是1x Perm Buffer II。避免在4℃以下培养样品,因为DMSO会结冰。
5. 如果0-稀释液和13-稀释液没有达到3的MFI倍数变化,与其制备13-稀释液,不如将15 μL 50稀释液和35 μL DMSO混合制成15-稀释液。
6. 较高浓度的FCB染料可能会改变相同荧光色素的MFI值,因为条形码样品的自发荧光增加。因此,当不需要MFI值时,首选较高的染料浓度或12个样品基质浓度用于表面标记染色(例如,免疫表型分型或T细胞亚群表征)。
7. 在培养后,最好将样品组合在已装满3 mL 1x Barcoding Wash Buffer的管中,因为在转移时,剩余未结合的染料会在较大体积的洗涤缓冲液中迅速稀释。
8. 为了去除在采集时可能与细胞发生反应的残留未结合染料,重复洗涤步骤两次,特别是如果没有在同一天采集条形码样品,则应及时进行洗涤。
9. 条形码“Combo”细胞可用于多达3种不同的抗体染色,通过转移100 μL细胞悬液在3个单独的管。相应地将管标记为“染色1”、“染色2”或“染色3”。如果只需要一次染色和/或采集更多的事件,可以将细胞重悬在200 μL 1x Barcoding Wash Buffer中,并直接将抗体添加到样品中。
10. 可以在门控策略的任意步骤进行反卷积,然而,由于两个原因,阳性细胞的百分比可能不同:
(a)如果在分析后期进行反卷积,则在识别条形码群体之前,所有门中不包括条形码细胞
(b)阳性细胞的百分比是使用一个给定的群体比例作为总群体(第一级细胞,100%)。例如,如果我们想知道CD4+/CD8+细胞的百分比: 如果我们先做反卷积,每个条形码人口是我们的上一级门所有每百分比将被计算。如果我们稍后做反卷积,我们首先识别出CD4+和CD8+细胞,这些亚群就成为我们的上一级群体。然后,如果我们对CD4+细胞进行反卷积,我们将识别出9个条形码群体,其百分比代表每个群体在CD4+亚群中的比例。
11. 在优化phosphoFlow与FCB联合应用的方案时,除了阳性细胞百分比和MFI外,还需要计算每个条形码人群的pSTATs的MFI倍数变化值(刺激样本的MFI /未刺激样本的MFI),并与匹配的对照组进行比较。
参考文献:
1. Finak G, Langweiler M, Jaimes M et al (2016) Standardizing flow cytometry immunopheno typing analysis from the human immunopheno typing consortium. Sci Rep 6:20686
2. Maecker HT, JP MC Jr, FOCIS Human Immunophenotyping Consortium et al (2010) A model for harmonizing flow cytometry in clinical trials. Nat Immunol 11(11):975–978
3. Maecker HT, McCoy JP, Nussenblatt R (2012) Standardizing immunophenotyping for the human immunology project. Nat Rev Immunol 12(3):191–200
4. Krutzik PO, Nolan GP (2006) Fluorescent cell barcoding in flow cytometry allows high throughput drug screening and signaling profiling. Nat Methods 3:361–368
5. Giudice V, Feng X, Kajigaya S, Young NS, Biancotto A (2017) Optimization and standardization of fluorescent cell barcoding for multiplexed flow cytometric phenotyping. Cytometry A 91:694–703
6. Lim CY, Owens NA, Wampler RD et al (2014) Succinimidyl ester surface chemistry: implications of the competition between aminolysis and hydrolysis on covalent protein immobilization. Langmuir 30(43):12868–12878